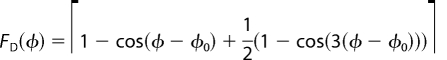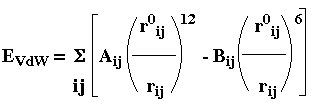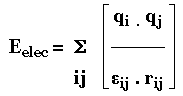| Mécanique moléculaire et modélisation moléculaire |
| Tweet |
|
|
1. Principe de la mécanique moléculaire 2. Le champ de force d'une protéine 3. Fonction de l'énergie potentielle d'une protéine |
4. Exemple : expression de l'énergie d'interactions entre atomes non liés 5. Liens Internet et références bibliographiques |
1. Principe de la mécanique moléculaire Les objectifs de la mécanique moléculaire et de la modélisation moléculaire sont, entre autres :
La mécanique moléculaire est une méthode empirique qui utilise un modèle mathématique et divers paramètres de potentiels : l'ensemble [modèle mathématique / paramètres de potentiels] s'appelle un champ de force. Les protéines sont constitués de centaines ou de milliers d'atomes et les seules méthodes de calculs pour des systèmes de cette taille sont les calculs de mécanique moléculaire. La mécanique quantique ne permet d'étudier des systèmes comportant quelques centaines d'atomes. |
|
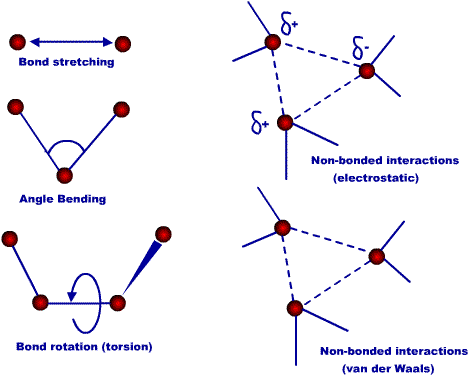
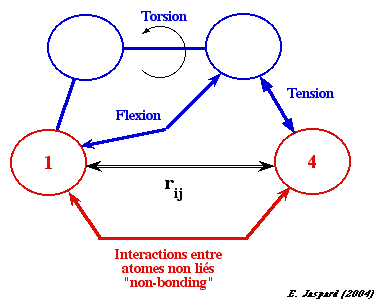
2. Le champ de force d'une protéine Les atomes sont traités comme des "balles en caoutchouc" de différentes tailles reliées entre elles par des "ressorts" (les liaisons) de différentes longueurs. Un champ de force est associé à chaque atome dans la protéine. Le schéma ci-dessous représente les cinq types de potentiels clés du champ de force :
Source : Tug Sezen - Folding@home
L'énergie totale d'une protéine est donnée par l'équation suivante : Etot = Estretch + Ebend + Etors + EVdW + Eelec
Autre formulation d'un champ de forces :
Source : Durrant & McCammon (2011) Selon la méthode de calcul utilisée pour le calcul de l'énergie, les structures moléculaires modélisées et leurs propriétés seront ou non exactes. Le choix du champs de force est donc à faire en se basant sur les résultats déjà obtenus dans la littérature concernant leurs applications aux systèmes moléculaires. Exemples de champ de force en mécanique moléculaire : |
|
Prix Nobel de chimie 2013 : Martin Karplus, Michael Levitt et Arieh Warshel "for the development of multiscale models for complex chemical systems".
Source : Brooks et al. (2009) Le canal KcsA K+ inséré dans membrane de phospholipide dipalmitoyl phosphatidylcholine.
============================================ VOIR A Python program to convert ParamChem CGenFF toppar stream file from CHARMM to GROMACS format. The comments section in the beginning of the program provides usage information. 2cgenff_charmm2gmx.py http://mackerell.umaryland.edu/charmm_ff.shtml#gromacs
charmm nonprofit/academic license |
|
3. Fonction de l'énergie potentielle d'une protéine Cette fonction tient compte des différents types d'interactions au sein d'une protéine. Ci-dessous : exemple d'une fonction PEF(R) ("Potential Energy Function") utilisée pour calculer la valeur de l'énergie potentielle d'une conformation d'une protéine.
Source : Jamros et al. (2010) r0, θ0, ξ0, et φ0 sont les valeurs déterminées à partir des structures cristallines. |
9. Modélisation moléculaire Bio.PDB (Package PDB) : Classes that deal with macromolecular crystal structures. Bio.SCOP (Package SCOP) : SCOP: Structural Classification of Proteins. FragBuilder: an efficient Python library to setup quantum chemistry calculations on peptides models PeptideBuilder: A simple Python library to generate model peptides |
| 5. Liens Internet et références bibliographiques |
|
Protein data bank Folding at home homology-modeling program ESyPred3D protein structure homology-modeling program DeepView (SwissPdb-Viewer) |
|
|
Brooks et al. (2009) "CHARMM: The Biomolecular Simulation Program" J. Comput. Chem. 30, 1545 - 1614 Durrant & McCammon (2011) "Molecular dynamics simulations and drug discovery" BMC Biol. 9, 71 |
![]()