| Bioinformatics analysis of the structure - function relationship of glutamate dehydrogenase |
| Tweet |
|
Jaspard E. (2006) "A computational analysis of the three isoforms of glutamate dehydrogenase reveals structural features of the isoform EC 1.4.1.4 supporting a key role in ammonium assimilation by plants" Biol. Direct. 1, 38 |
|
Préambule On peut considérer la première réaction d'assimilation de l'azote (sous forme d'ammoniac) par la glutamate déshydrogénase (GDH) comme un point d'entrée dans le métabolisme azoté. L'atome d'azote est à l'origine de la fonction α-aminée des acides aminés selon la réaction : NH3+ + α-cétoglutarate + NAD(P)H + H+ <=> glutamate + NAD(P)+ Il existe trois isoformes de GDH :
La GDH4 joue peut-être un rôle clé dans l'assimilation de l'azote. Or ce rôle n'a pas encore été démontré, notamment chez les plantes. Par ailleurs, on ne dispose d'aucune information concernant la structure de la GDH4. |
In term of taxonomy, the closest organism to higher plants (Eukaryota, Viridiplantae, Streptophyta) from which sequences of glutamate dehydrogenase EC 1.4.1.4 are known is an algae, Chlorella sorokiniana (Eukaryota, Viridiplantae, Chlorophyta). Thus, the two sequences of this isoform from Chlorella sorokiniana were taken as the reference (Ref) subset in this study. Classification of the 116 selected full GDHs amino acid sequences 116 non-redundant full GDH sequences were obtained from 83 organisms representing the three domains, Archaea, Bacteria and Eukaryota. These sequences were classified in 15 different subsets using the following criteria :
The following table gives the number of amino acid sequences of GDH for each subset (letter). The length range of the polypeptide chains are : L1 = [411 : 470] - L2 = [503 : 558] - L3 = [1029 : 1106] - L4 = [1607 : 1651] |
| EC number | Viridiplantae | not Viridiplantae | total | ||||||
| L1 | L2 | L3 | L4 | L1 | L2 | L3 | L4 | ||
| 1.4.1.2 | 9 (A) | 7 (B) | 1 (C) | 17 | |||||
| 1.4.1.3 | 1 (D) | 6 (E) | 7 (F) | 14 | |||||
| 1.4.1.4 | 2 (Ref) | 15 (G) | 17 | ||||||
| not classified | 6 (H) | 13 (I1) 18 (I2) 14 (I3) | 5 (J) | 5 (K) | 7 (L) | 68 | |||
| total | 16 | 2 | 73 | 12 | 5 | 8 | 116 | ||
The sequences of each subset were further aligned to obtain the 15 full consensus amino acids sequences. |
|
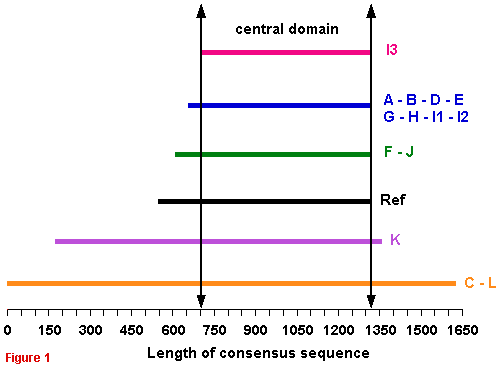
Comparison of the 15 full consensus GDHs sequences Unzip (untar) the following text file containing the 15 full consensus sequences in FASTA format : FullConsSeq.zip and FullConsSeq.tar The schematic alignment of the full consensus sequences shows that GDH subunit is constituted of two or three regions :
|
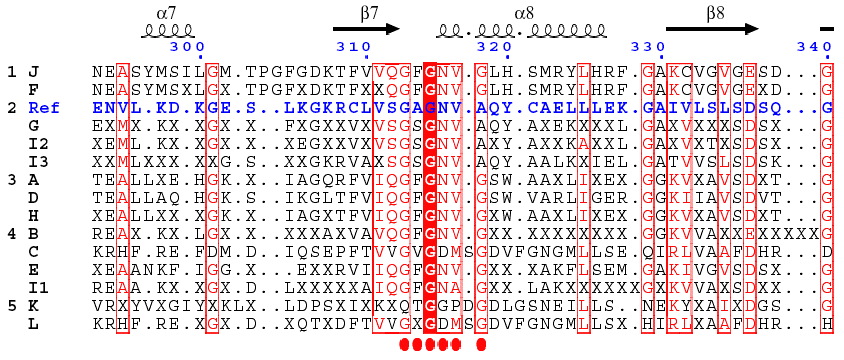
Analysis of the central domain of GDH : the dinucleotide-binding motif A βαβ fold is found in the NAD(P)H-binding subdomain (β7 - α8 - β8). This Rossmann fold begins with the motif G313AGNVA318 in the case of Ref. However, the alignment indicates that the actual motives could be more complex. Such a higher complexity of the signature for the NAD(P)H-binding motif allows to discriminate more precisely the three isoforms. The figure was built using ESpript.
|
|
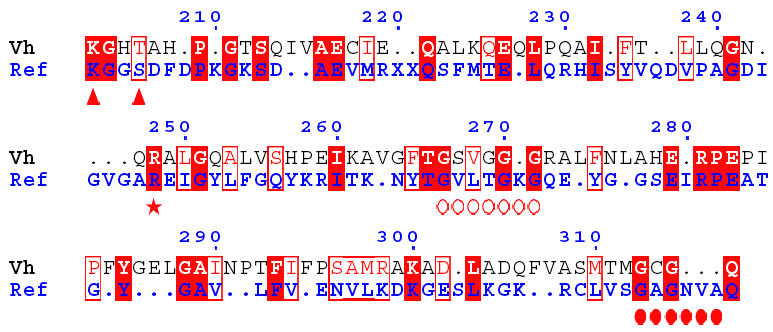
Search of a second NAD(P)H-binding site Aldehyde dehydrogenase from Vibrio harveyi is one of the most NADP-specific. The alignment of GDH from Ref and aldehyde DH shows that :
Therefore, the latter is likely a second nucleotide-binding motif specific of GDH4. |
|
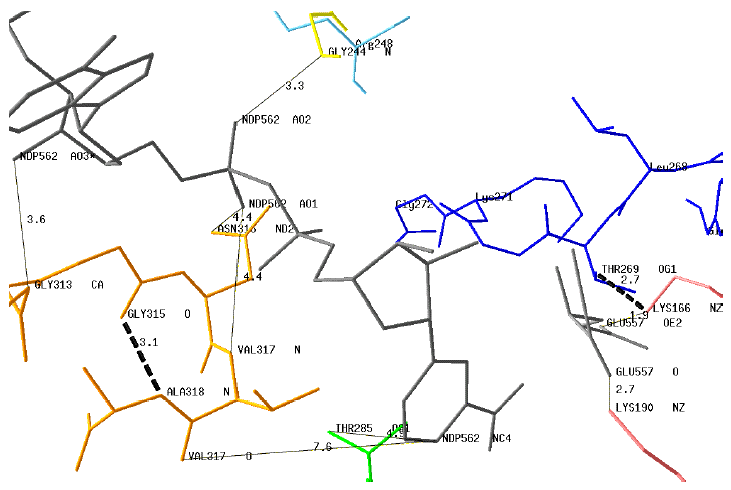
Modelisation of the dinucleotide-binding motives and key residues of GDH4 with NADPH (NDP562) and Glu A theoretical 3D structure of GDH4 from Ref was generated with the homology-modeling program ESyPred3D using as the template the structure of bovine GDH3 (PDB # 1HWZ). The modelisation and the drawing of a putative structure of GDH4 was performed with the protein structure homology-modeling program DeepView (SwissPdb-Viewer v. 3.7). Some interactions (plain lines) between the motif G313AGNVA318 or key residues and the coenzyme are indicated : NDP562AO3 - Gly313CA; NDP562AO1 - Asn316ND2; NDP562AO1 - Val317N; NDP562AO2 - Gly244N; NDP562NC4 - Thr285OG1
The distances between the protonated carbon atom of the nicotinamide moiety (NDP562NC4) are too long for direct interactions with the motif G313AGNVA318. However, this motif is stabilized by an internal H-bond Gly315O - Ala318N (dotted line). Two distances (Glu557OE2 - Lys166NZ and Glu557O - Lys190NZ) are compatible with H-bond interactions between the enzyme and Glu. The position of the motif G266VLTGKG272 is shown with the potential H-bond Lys166NZ - Thr269OG1. |
| ESpript : Gouet, P., Courcelle, E., Stuart, D. I. & Metoz, F. (1999) "ESPript : analysis of multiple sequence alignments in PostScript" Bioinformatics 15, 305 - 308 | ESpript |
| ESyPred3D : Lambert, C., Leonard, N., De Bolle, X. & Depiereux, E. (2002) "ESyPred3D : Prediction of proteins 3D structures" Bioinformatics 18, 1250 - 1256 | EsyPred3D |
| Swiss-PdbViewer : Guex, N. & Peitsch M. C. (1997) "SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling" Electrophoresis 18, 2714 - 2723 | DeepView |
![]()