ENZYMOLOGIE
Historique - site actif - complexe enzyme/substrat - état de transition - protéases
ENZYMOLOGIE Historique - site actif - complexe enzyme/substrat - état de transition - protéases |
Chargement de la page : "Historique - site actif - complexe enzyme/substrat - état de transition - protéases"
|
1. Historique 2. Spécificité de l'association [protéine - ligand] - Notion de site actif - Classes d'enzymes (E.C. X.X.X.X) 3. Le complexe enzyme - substrat 4. Etat de transition, énergie libre d'activation et énergie libre de la réaction enzymatique
|
5. Les effets de la sur-population intracellulaire des macromolécules ("Macromolecular crowding effects") 6. Illustration des protéases
7. Mécanisme catalytique des protéases à sérine Annexe : distinction entre concentrations à l'équilibre et à l'état stationnaire |
Les organismes vivants sont le siège d'un grand nombre de réactions biochimiques très diverses. Ces réactions s'effectuent dans des conditions où, normalement, elles ne pourraient se faire. Si elles ont lieu, c'est parce qu'elles sont catalysées par des macromolécules biologiques : les enzymes. Le pouvoir de catalyse des enzymes est lié, entre autre, à la très haute spécificité de reconnaissance des molécules sur lesquelles elles agissent.
|
| 1. Historique. Il est difficile de situer exactement la découverte de la notion d'enzyme et surtout d'enzyme en tant que seul catalyseur des réactions chimiques qui se déroulent dans les organismes vivants. 1783 : Lazzaro Spallanzani a rapporté que la viande est liquéfiée par un extrait gastrique. Il a noté également que la température a un grand effet. 1814 : Constantin Kirchhoff observa qu'un composant "glutineux" (comme il l'a appelé à l'époque) de blé convertit l'amidon en sucre. 1833 : La première découverte d'une enzyme est d'habitude attribuée à Anselme Payen et Jean-François Persoz [Payen A & Perzoz J (1833) "Mémoire sur la diastase, les principaux produits de ses réactions et leurs applications aux arts industriels" Annales de la chimie et de la physique 53, 73-92]. Ils ont traité un extrait aqueux de malt à l'éthanol puis précipité une substance labile à la chaleur qui hydrolyse l'amidon. Ils ont appelé cette fraction "diastase" ("séparation" en Grec) puisque cette fraction sépare le sucre soluble de l'amidon insoluble. On sait maintenant que cette préparation était une solution non purifiée d'amylase. 1834 : Theodor Schwann a obtenu le premier un agent actif d'origine animale (la pepsine) qu'il a partiellement purifiée en traitant la paroi stomacale par l'acide. Il est important de souligner qu'à l'époque les premières observations d'activité enzymatique ont précédé une notion claire et précise de la catalyse. Le concept de catalyse provient de l'observation de l'action de la diastase et de la pepsine parallèlement à celle de la levure pendant la fermentation : dans tous les cas, une "substance était changée en une autre" sous l'influence d'un agent actif : le catalyseur. A l'époque, la levure n'était pas encore considérée comme une cellule vivante. 1838 : Charles Cagniard de Latour montra que le processus de fermentation est dû à des organismes vivants. 1858 à 1871 : Les travaux de Louis Pasteur confirmèrent cette idée. Pasteur émit l'hypothèse révolutionnaire que les changements chimiques lors de la fermentation résultaient des processus de la vie des micro-organismes impliqués dans la fermentation. A l'opposé, Justus von Liebig privilégiait une théorie purement chimique : un "ferment" était une substance chimique produite par un organisme en décomposition et les atomes de ce ferment étaient supposés en mouvement incessant. Cet état d'agitation élevé était transmis aux atomes de la molécule de sucre (substrat du ferment) dont les éléments devaient être maintenus par des forces faibles. Il en résultait une scission du sucre en CO2 et éthanol dont les liaisons étaient plus fortes. 1860 : Marcellin Berthelot fit macérer de la levure et obtint une fraction précipitable à l'alcool capable de convertir le sucrose en glucose plus fructose. Il conclut que l'invertase (nom qu'il donna à l'agent actif de cet extrait) était l'un des multiples ferments présents dans la levure. L'invertase est la β-fructofuranosidase (E.C. 3.2.1.26). 1876 - 1877 : Wilhelm Kühne a découvert la trypsine dans le liquide pancréatique de l'intestin de boeuf au cours de ses études sur la digestion intermédiaire des protéines dans le tractus gastro-intestinal. Il a conclu que la trypsine était initialement inactive puis convertie en sa forme active. Cette observation est à la base de la notion de précurseur inactif des protéases que l'on appelle zymogène et de l'activation de ce zymogène par (auto)protéolyse. 1878 : Wilhelm Kühne proposa le nom d'enzyme ("En-" et "zumê", signifiant "dans le levain") pour qualifier ces ferments. L'addition du suffixe "ase" au nom du substrat d'une enzyme pour dénommer cette enzyme fût proposé par Emile Duclaux en 1898. 1896 : Un chimiste allemand, Martin Hahn, tentait d'isoler des protéines de levure (Saccharomyces cerevisiae) en broyant ces levures dans un mortier avec du sable fin et de terre de diatomées. Cependant, cette préparation d'extrait de levure se conservait mal. Hans Buchner et Eduard Buchner
s'intéressaient aussi aux extraits de levure dans un but thérapeutique.
Ces extraits étant destinés à l'homme ne pouvaient contenir des
bactéricides. Se souvenant que le
sucre permet de conserver les fruits, Hans Buchner, proposa d'ajouter
du saccharose à l'extrait de levure pour le conserver. En publiant ses observations, Eduard Buchner concluait : "La complexité de la levure n'est pas indispensable au déroulement de ce processus. Le ferment actif du jus n'est probablement qu'une substance dissoute, sans aucun doute une protéine : nous la baptiserons zymase". Pour cette découverte, Eduard Buchner reçut le Prix Nobel en 1907. On sait maintenant que la "zymase" des extraits de levure est en fait un mélange d'enzymes dont l'ensemble catalyse les réactions de la glycolyse. 1897 : La même année, Gabriel Bertrand observa que certaines enzymes requièrent des facteurs dialysables pour leur activité. Il les nomma coenzymes. |
|
Au début du 20è siècle, de gros travaux furent entrepris pour purifier des enzymes et surtout décrire leur activité catalytique en termes mathématiques. 1902 : Victor Henri et Adrian Brown suggérèrent indépendamment que la formation d'un complexe enzyme - substrat est un intermédiaire obligatoire de la réaction enzymatique.
Victor Henri fût donc le premier à décrire l'équation mathématique reliant l'effet de la concentration du substrat à la vitesse de catalyse. Article : V. Henri (1902) "Théorie générale de l'action de quelques diastases" C. R. Hebd. Séances Acad. Sci. 135, 916 - 919 1913 : En étudiant les propriétés catalytiques de l'invertase (sucrose => fructose + glucose), Maud Menten et Leonor Michaelis redécouvrirent l'équation de Victor Henri et établirent la relation connue sous le nom d'équation de Henri - Michaelis - Menten. Ils en interprétèrent correctement la signification des constantes . Le point important est que l'obtention de cette équation repose sur l'hypothèse qu'il s'établit un équilibre rapide entre les concentrations de l'enzyme, du substrat et du complexe enzyme - substrat (E + S <=> ES). Articles :
1925 : George Briggs et John Haldane généralisèrent l'équation précédente en introduisant le concept d'état stationnaire pour la concentration du complexe enzyme - substrat. Article : Briggs & Haldane (1925) "A note on the kinetics of enzyme action" Biochem J. 19, 338 - 339 |
|
Le fait que les enzymes sont des protéines ne fût accepté qu'à partir de la fin des années 20. De nouvelles techniques chimiques et physiques furent employées pour analyser la structure des protéines. 1926 : James Sumner (Prix Nobel en 1946) cristallisât l'uréase. Années 30 : John Northrop (Prix Nobel en 1946) et ses collaborateurs cristallisèrent le pepsinogène, la pepsine et plusieurs isoformes de la trypsine et de la chymotrypsine et démontrèrent la pureté des cristaux obtenus. Années 40 et 50 : Des centaines d'enzymes furent purifiées et cristallisées permettant ainsi l'élucidation de dizaines de voies métaboliques. 1955 : Frédéric Sanger (1er Prix Nobel en 1958) publia la séquence complète en acides aminés d'une petite protéine : l'insuline (masse molaire 6000 Da). 1957 : La première structure cristallographique d'une protéine (la myoglobine), déduite de la diffraction des rayons X, fût obtenue par John Kendrew (Prix Nobel en 1962 avec Max Perutz). Années 60 : La première séquence d'une enzyme (la ribonucléase, masse molaire 13700 Da) fût publiée en 1960 et la première synthèse chimique (également de la ribonucléase) fût obtenue en 1969. Les biochimistes focalisèrent alors sur le mécanisme de l'activité enzymatique et son mode de régulation. 1958 : Daniel Koshland a proposé le modèle de l'ajustement induit. Le substrat induit un changement conformationnel du site actif de l'enzyme. 1961 : Christian Anfinsen (Prix Nobel en 1972) et ses collaborateurs ont montré que, pour un grand nombre de protéines, c'est la séquence en acides aminés qui détermine le repliement de la chaîne polypeptidique dans son unique conformation native donc active. Cette conformation est la plus stable parce qu'elle a une énergie libre minimale. 1963 : William Wallace Cleland proposa une procédure claire et uniforme pour écrire les équations des cinétiques des systèmes enzymatiques à plusieurs substrats. 1965 : Jacques Monod (Prix Nobel en 1965), Jeffries Wyman et Jean-Pierre Changeux proposèrent un modèle cinétique (modèle MWC) pour les enzymes allostériques (enzymes dont la courbe de vitesse en fonction de la concentration en substrat est une sigmoïdale et non une hyperbole). 1966 : Daniel Koshland, Georges Nemethy et D. Filmer généralisèrent le modèle précédent (modèle KNF) en incluant la notion d'ajustement induit proposé par Koshland. |
Années 80 à nos jours L'hypothèse de Chritian Anfinsen, vérifiée pour un grand nombre de protéines (notamment dites "globulaires") est restée longtemps l'un des dogmes de la biologie. Cependant, à partir des années 1980, 2 autres processus ont été mis en évidence : - les protéines chaperones (Ron Laskey - 1978; John Ellis - 1987) aident au repliement de certaines protéines ou les maintiennent dans la conformation native quand la cellule est soumise à certains stress. Parmi les chaperonnes, on peut citer :
- les protéines intrinsèquement non structurées : elles n'ont pas de structure tridimensionnelle à l'état libre et n'acquière leur structure repliée, donc fonctionnelle, que quand elles interagissent avec leurs partenaires cellulaires. Des approches plus récentes sont venues compléter les connaissances acquises pendant plus d'un siècle :
|
Avènement des méthodes liées à l'intelligence artificielle Enfin, l'avènement des méthodes d'apprentissage profond (domaine de l'intelligence artificielle) ouvre de nouvelles perspectives pour l'analyse de la relation structure-fonction des protéines en général et des enzymes en particulier. Le prix Nobel de chimie 2024 a ainsi récompensé la conception informatique des protéines et la prédiction de la structure des protéines (à partir de leur séquence en acides aminés) avec des outils issus de l'apprentissage profond : Rosetta et AlphaFold, respectivement. Par ailleurs, les performances croissantes des grands modèles de langage naturel appliqués aux séquences de protéines génèrent des prédictions d'une précision inégalée. |
| Quelques bases de données dédiées aux enzymes | |
| Enzyme Nomenclature | Protein Data Bank |
| ExPASy (Expert Protein Analysis System) | BRENDA |
| ENZYME - Enzyme nomenclature database | ExplorEnz - The Enzyme Database |
| UniProt | KEGG Database |
Rhea : base de données de réactions chimiques et de transport d'intérêt biologique, conçue pour l'annotation fonctionnelle des enzymes et des transporteurs dans UniProtKB.
Bansal et al. (2021) "Rhea, the reaction knowledgebase in 2022" Nucleic Acids Res. 50, D693 - D700 |
|
2. Spécificité de l'association [protéine - ligand] - Notion de site actif a. Toutes les protéines se replient dans une conformation dite native et c'est dans cette conformation qu'elles acquièrent leur activité biologique (leur pouvoir de catalyseur dans le cas des enzymes). Ce repliement aboutit à une structure tridimensionnelle fonctionnelle unique de la protéine. Cas particulier : les protéines dites intrinsèquement non structurées n'adoptent leur conformation native que transitoirement (tout ou partie de la chaîne polypeptidique étant concernée) lors de l'interaction avec leur(s) ligand(s). |
b. Cette structure globale de la macromolécule permet à une région particulière d'adopter elle aussi une structure spatiale qui est reconnue par le ligand spécifique de la protéine (et, le cas échéant, par un petit nombre de molécules dont la structure est proche de celle du ligand). Dans le cas des enzymes, cette région particulière s'appelle le site actif et elle est généralement enfouie au sein de la structure repliée de l'enzyme. Le site actif a deux fonctions :
On peut citer divers types d'association entre une protéine et un ligand :
Si elle est réversible, l'association protéine - ligand correspond à l'équilibre suivant : k1 P + L <======> PL k-1 k1 : constante de vitesse microscopique du second ordre (réaction bimoléculaire). Unités : mol-1.L.s-1 ou M-1.s-1 k-1 : constante de vitesse microscopique du premier ordre (réaction monomoléculaire). Unités : s-1 On définit la constante de dissociation (constante macroscopique) qui lie la concentration des composés impliqués dans la réaction :
|
| L'énergie libre de Gibbs de formation des complexes [ΔGf = - RT Ln (1/Kd)] est très variable comme le montre le tableau suivant : | |||
| Protéine | Ligand | Kdissociation (M) | ΔG (kcal.mol-1) |
| Carboxypeptidase | β-phénylpropionate | 5 10-3 | 3,2 |
| Trypsine | butylamine | 10-3 | 4,2 |
| Phosphofructokinase (2 conformations) | adénosine diphosphate (ADP) | 10-3 - 2 10-5 | 4,2 - 6,5 |
| Homosérine déshydrogénase | NADPH | 3 10-7 | 9 |
| Méthionine - ARNt synthétase | méthionyl - adénylate | 2 10-9 | 12 |
| Histone | ADN | 10-11 | 15 |
| Apohémoglobine | hème | 10-13 | 18 |
| Source : "Biochimie" Chapeville, Clauser et al. (1974) Ed. Hermann | |||
d. Les enzymes ne fixent pas seulement un ligand (qui dans ce cas s'appelle un substrat). Elles le transforment en un produit lors d'une réaction chimique. Certains acides aminés du site actif ont donc pour fonction, non pas de fixer le substrat, mais de fournir les groupements chimiques nécessaires à la réaction catalysée par l'enzyme. Dans le cas des enzymes, on distingue donc au sein du site actif :
|
c. Le site actif est constitué d'un petit nombre d'acides aminés qui le plus souvent ne sont pas contigus dans l'enchaînement de la chaîne polypeptidique.
Source : Enzyme Mechanisms Ces acides aminés sont caractérisés par une chaîne latérale dont à la fois la nature chimique (groupement ionisable ou polarisable) et la structure (orientation et encombrement stérique) sont spécifiquement adaptés à la reconnaissance du (ou des) ligand(s). La stéréochimie qui résulte de cet agencement unique des résidus d'acides aminés qui constituent le site actif est la cause de la stéréospécificité de reconnaissance entre ces acides aminés et le (ou les) ligand(s). |
|
Les différentes classes d'enzymes
La classification établie par la commission des enzymes de l'Union Internationale de Biochimie et de Biologie Moléculaire (sigle anglais "IUBMB") a été établie sur des critères de spécificité. La nomenclature des enzymes s'écrit de manière générale sous la forme : E.C. X.X.X.X (E.C. : "Enzyme Commission"). La liste de toutes les enzymes, leur nomenclature et leur identifiant EC est disponible dans la base de données : Enzyme Nomenclature and Classification of Enzyme-Catalysed Reactions |
| Le premier "X" correspond aux 6 types de réactions catalysées par les enzymes. | 1er X | Réaction catalysée | exemple de coenzyme impliqué |
| X = 1 : oxydoréductases (E. C. 1.X.X.X) | oxydoréduction | NAD(P)+ | |
| X = 2 : transférases (E. C. 2.X.X.X) | transfert de groupes | phosphate de pyridoxal | |
| X = 3 : hydrolases (E. C. 3.X.X.X) | hydrolyse | aucun | |
| X = 4 : lyases (E. C. 4.X.X.X) | addition de groupe à des atomes engagés dans des doubles liaisons | pyrophosphate de thiamine | |
| X = 5 : isomérases (E. C. 5.X.X.X) | isomérisation (de position de groupe ou de fonction) | phosphate de pyridoxal | |
| X = 6 : ligases (E. C. 6.X.X.X) | condensation de deux molécules | ATP | |
| X = 7 : translocases (E. C. 7.X.X.X) | transfert d'un "côté 1" à un "côté 2" | aucun | |
Une nouvelle classe E.C. = 7 : translocases, a été ajoutée en 2018. |
|||
| Considérons le groupe 1
des oxydoréductases : le 2ème "X" permet un classement supplémentaire en fonction de la nature du groupe du donneur d'électrons sur lequel l'enzyme agit. |
2ème X | l'enzyme agit sur : |
| X = 1 : E. C. 1.1.X.X | le groupe CH-OH du donneur d'électrons | |
| X = 2 : E. C. 1.2.X.X | la fonction aldéhyde ou oxo du donneur d'électrons | |
| X = 3 : E. C. 1.3.X.X | le groupe CH-CH du donneur d'électrons | |
| etc ... | ||
| X = 19 : E. C. 1.19.X.X | la flavodoxine réduite | |
| X = 97 : E. C. 1.97.X.X | autres oxydoréductases | |
|
Considérons le groupe 1.4
des oxydoréductases qui agissent sur le groupe CH-NH2 du donneur
d'électrons : le 3ème "X" permet un classement supplémentaire en fonction de la nature du groupe accepteur d'électrons sur lequel l'enzyme agit. |
3ème X | l'accepteur d'électrons est: |
| X = 1 : E. C. 1.4.1.X | NAD+ ou NADP+ | |
| X = 2 : E. C. 1.4.2.X | un cytochrome | |
| X = 3 : E. C. 1.4.3.X | l'oxygène | |
| X = 4 : E. C. 1.4.4.X | un groupe disulfure | |
| X = 7 : E. C. 1.4.7.X | une protéine fer - soufre | |
| X = 99 : E. C. 1.4.99.X | autres accepteurs | |
| Remarque : il n'y a pas de classe E. C. 1.4.5.X et E. C. 1.4.6.X | ||
|
Considérons le groupe
1.4.1 des oxydoréductases qui utilisent le NAD+ ou le NADP+ comme
accepteur d'électrons : le 4ème "X" permet un classement supplémentaire en fonction du substrat sur lequel l'enzyme agit. |
4ème X | Nom de l'enzyme |
| X = 1 : E. C. 1.4.1.1 | alanine déshydrogénase | |
| X = 2 : E. C. 1.4.1.2 | glutamate déshydrogénase | |
| X = 3 : E. C. 1.4.1.3 | glutamate déshydrogénase (NAD(P)+) | |
| X = 4 : E. C. 1.4.1.4 | glutamate déshydrogénase (NAD(P)+) | |
| etc ... | ||
| X = 19 : E. C. 1.4.1.19 | tryptophane déshydrogénase | |
| X = 20 : E. C. 1.4.1.20 | phénylalanine déshydrogénase | |
L'exemple de la glutamate déshydrogénase (E. C. 1.4.1.2, E. C. 1.4.1.3 et E. C. 1.4.1.4) met en évidence la notion d'isoenzymes ou d'isoformes de la "même" enzyme. Voir la base de données : ExplorEnz - The Enzyme Database Voir l'article : Sorokina et al. (2014) "Profiling the orphan enzymes" Biology Direct 9, 10 |
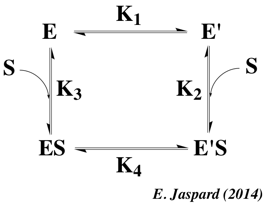
3. Le complexe enzyme - substrat Le schéma réactionnel minimal pour une réaction enzymatique à un substrat S et un produit P est : E + S <=> ES <=> E + P La formation initiale d'un complexe [enzyme - substrat] ES ou complexe de Michaelis-Menten-Henri, NON covalent, fût suggérée d'après les observations suivantes :
L'hypothèse "clé -serrure", bien qu'extrêmement satisfaisante, ne peut cependant pas rendre compte de certaines observations :
|
Nature des forces dans les associations [enzyme - substrat]
|
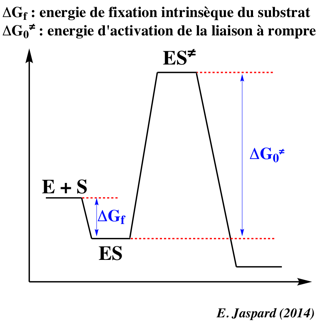
Variation d'énergie libre de Gibbs de formation du complexe [enzyme - substrat] Si KS est la constante de dissociation du complexe [enzyme - substrat], l'énergie libre de Gibbs de formation du complexe est : ΔGf = - RT Ln (1/KS).
Les valeurs de ΔG pour la formation du complexe de Michaelis-Menten-Henri sont généralement assez faibles et toujours négatives. En d'autres termes, la formation de ce premier complexe s'accompagne d'une diminution de l'énergie libre de Gibbs du système : c'est un processus spontané. |
| Exemples de valeurs de paramètres thermodynamiques de l'association [enzyme - substrat] ou [enzyme - inhibiteur] | ||||
| Enzyme | Substrat ou inhibiteur | ΔG (kcal.mol-1) | ΔH (kcal.mol-1) | ΔS (u. e.) |
| Chymotrypsine | méthyl hydrocinnamate |
- 1,9 |
- 5,3 |
- 11,4 |
| Carboxypeptidase | carboxybenzoxy-L-tryptophane carboxybenzoxy glycyl-L-phénylalanine carboxybenzoxy glycyl-L-tryptophane |
- 3,4 - 2,5 - 3,4 |
5 - 0,4 0 |
- 28,0 - 7,0 - 11,5 |
| Pepsine | carboxybenzoxy-L-glutamyl-L-tyrosine éthyl ester |
- 4,7 - 4,3 |
- 1,4 - 3,0 |
20,6 24,4 |
| Uréase | urée | - 3,2 | - 2,9 | 0,9 |
| ATPase | ATP | - 7,5 | 8 | 52 |
| Source : "Enzymologie moléculaire et cellulaire" J. Yon-Kahn & G. Hervé (2005) Coll. Grenoble Sciences (EDP Sciences) | ||||
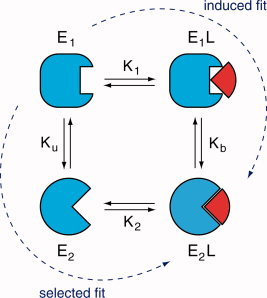
Diverses théories ont été proposées pour expliquer les modifications structurales du substrat et/ou de l'enzyme lors de leur association. a. Henry Eyring et Rufus Lumry (1954) ont proposé le modèle appelé "rack" (traduction : chevalet de torture ; tourmenter). C'est le substrat qui subit un changement conformationnel, la structure de l'enzyme étant considérée comme rigide. Le résultat est un accroissement de la labilité des liaisons à rompre dans la molécule de substrat puisque celui-ci, bien que fixé de manière lâche, subit des tensions dans ses liaisons qui l'amènent dans une configuration proche de celle de l'état de transition. b. En 1958, Daniel Koshland a proposé le modèle dynamique de l'ajustement induit ("induced fit"). Le substrat induit un changement conformationnel du site actif de l'enzyme.
Dans le modèle de l'ajustement induit (figure ci-dessous) :
Adapté de "Enzymologie moléculaire et cellulaire" Yon-Kahn & Hervé (2005)
Exemples de mécanismes en faveur de changements conformationnels de l'enzyme consécutifs à la fixation d'un ligand (substrat ou inhibiteur) :
c. En 1966, Williams Jenks a proposé le modèle appelé "strain" (tension - déformation). C'est une combinaison des deux théories qui précèdent. Les changements de conformation de l'enzyme entraînent des contraintes dans la structure du substrat. |
d. Ajustement induit ou sélection induite ? Une question centrale est de savoir :
La figure ci-dessous illustre les deux modèles :
Source : Weikl & Deuster (2009) Proteins 75, 104-110
|
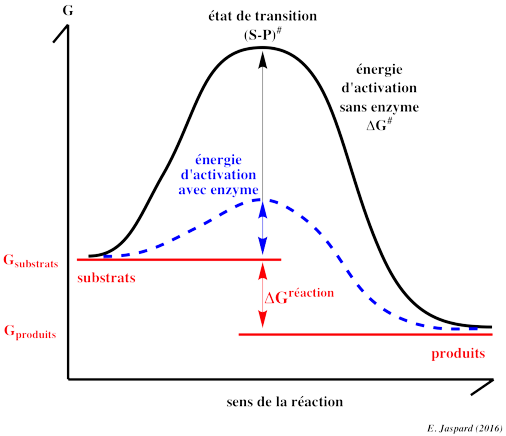
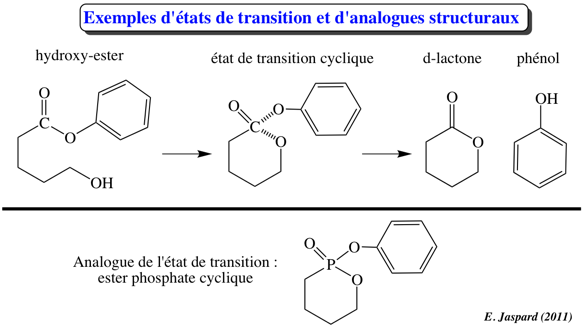
4. Etat de transition, énergie libre d'activation et énergie libre de la réaction enzymatique D'une part, la différence d'énergie libre de Gibbs entre le(s) produit(s) et le(s substrat(s) d'une réaction est la variation d'énergie libre de Gibbs de cette réaction : ΔGréaction. Par ailleurs, un substrat et un produit d'une réaction enzymatique sont caractérisés par des liaisons chimiques.
L'énergie requise pour que la réaction ait lieu (l'énergie nécessaire pour que les liaisons du substrat soient rompues) s'appelle l'énergie libre d'activation : ΔG#.
|
Henry Eyring, Meredith Evans et Michael Polanyi ont développé la théorie de l'état de transition en 1935 : Quand toute l'énergie d'activation est absorbée, la molécule de substrat est dans l'état de transition (les angles et les longueurs des liaisons chimiques du substrat sont distordues).
Seule l'intervention de catalyseurs biologiques, les enzymes, permet que les macromolécules franchissent la barrière d'activation dans un laps de temps compatible avec les processus biologiques. En effet, une enzyme augmente la vitesse de la réaction en abaissant l'énergie d'activation : à la même température, les substrats franchissent plus facilement et donc plus fréquemment la barrière d'activation. En conclusion :
Dans les conditions de la vie cellulaire, mais sans enzyme, peu de molécule peuvent franchir la barrière d'activation dans un laps de temps compatible avec les processus biologiques. Par exemple, le demi-temps de désamination de l'adénosine à 20°C et à pH7 est d'environ 20.000 ans. |
1er facteur : les deux grands types chimiques de mécanismes catalytiques α. La catalyse acide-base générale : c'est le mécanisme le plus courant de la catalyse enzymatique. L'accélération de la réaction résulte du transfert d'un proton. Par exemple, ce proton peut provenir du couple [imidazole / imidazolium] de la chaîne latérale de l'histidine puisqu'elle a un pKa situé entre 6 et 7 et constitue au pH physiologique un groupe [donneur / accepteur] de proton. L'activité catalytique des enzymes de ce type est largement influencée par le pH. Exemples de groupes fonctionnels d'acides aminés qui peuvent agir en tant que catalyseur acide/base :
β. La catalyse par covalence (aussi appelée catalyse nucléophile) : elle concerne essentiellement les réactions enzymatiques où plusieurs substrats sont impliqués (environ 20% des enzymes et toutes les enzymes qui catalysent un mécanisme à 2 substrats de type "ping-pong"). Une partie du premier substrat est transférée à l'enzyme via la formation d'une liaison covalente puis cette partie du substrat est transférée au second substrat. |
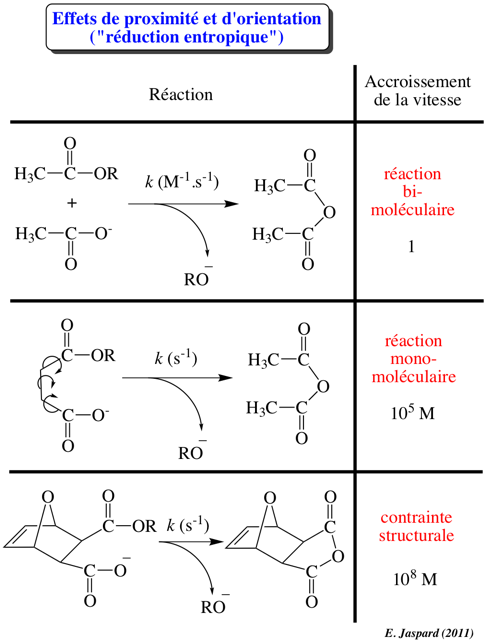
2ème facteur : l'effet de voisinage
La fixation d'une molécule de substrat au site actif d'une enzyme augmente la concentration effective du substrat par rapport à sa concentration à l'état libre en solution. Les molécules ne sont plus assujetties au hasard de collisions suffisamment énergétiques. Cet accroissement de concentration augmente la fréquence de formation de l'état de transition. |
3ème facteur : la fixation préférentielle de l'état de transition
L'enzyme fixe plus fortement l'état de transition que le substrat ou le produit pour des raisons de contrainte ou de distorsion. L'état de transition s'ajuste mieux que le substrat car il établit davantage de contacts avec les résidus d'acides aminés du site actif. C'est trés probablement l'enzyme qui subit des changements de conformation (voir ajustement induit) car il est peu probable qu'il y ait suffisamment d'énergie dans la fixation du substrat pour en contraindre la structure. |
4ème facteur : la stabilisation de l'état de transition Quand le complexe [enzyme - substrat] est formé, il est en équilibre avec sa forme activée, c'est-à-dire le complexe [enzyme - état de transition] ou ES#. La stabilisation de l'état de transition déplace l'équilibre en faveur de ES# et en même temps abaisse l'énergie d'activation. Il est important de noter que ce phénomène exige une fixation lâche du substrat (attention : pas une fixation lâche de l'état de transition) à l'enzyme car une fixation trop forte irait à l'encontre de la catalyse. L'enzyme doit donc maintenir le substrat en place tant que l'état de transition n'est pas atteint mais sans l'immobiliser. Si tel était le cas, le complexe ES formé serait trop stable pour atteindre sa configuration de l'état de transition (ES#). |
| 5ème facteur : la catalyse électrostatique
Une protéine possède de nombreuses charges ainsi que des dipôles qui créent des champs électrostatiques dont l'intensité est très variable selon les régions de la protéine. De plus ces charges fluctuent en fonction des changements de conformation de la protéine. Quand le substrat se fixe à l'enzyme, les molécules d'eau sont de manière générale "exclues" du site actif (effet de désolvatation).
Ainsi, les champs électrostatiques créés par la protéine au niveau du site actif sont complémentaires de la distribution des charges du substrat dans l'état de transition. Un cas particulier est l'eau quand elle agit comme substrat (exemple des protéases à sérine). L'ensemble de tous les facteurs énumérés rend compte des valeurs d'accélération couramment observées avec les enzymes : |
| Enzyme | Vitesse non enzymatique (s-1) | Vitesse enzymatique (s-1) | Facteur d'accroissement |
| Chymotrypsine | 4 10-9 | 4 10-2 | 107 |
| Lysozyme | 3 10-9 | 5 10-1 | 2 108 |
| Triose phosphate isomérase | 6 10-7 | 2 103 | 3 109 |
| Fumarase | 2 10-8 | 2 103 | 1011 |
| Uréase | 3 10-10 | 3 104 | 1014 |
| Désaminase d'adénosine | 10-12 | 102 | 1014 |
| Phosphatase alcaline | 10-15 | 102 | 1017 |
| Source : "Principes de Biochimie" Horton, Moran, Ochs, Rawn, Scrimgeour (1994), Ed. DeBoecK Universités | |||
5. Les effets de la sur-population intracellulaire des macromolécules ("Macromolecular crowding effects") Les conditions "idéales" d'étude des cinétiques enzymatiques ne reflètent pas la réalité cellulaire. Le milieu intracellulaire n'est pas rempli que d'eau et d'ions : il contient une très grande variété de petites molécules et de macromolécules (protéines, acides nucléiques, oligosaccharides, acides gras, métabolites, ...). La concentration des macromolécules peut varier de 200 à 400 mg/ml selon l'organisme et les types de cellules :
Source : McGuffee & Elcock (2010)
Le volume du cytoplasme de Escherichia coli correspond à 600 cubes de 100 nm d'arête. Dans un de ces cubes, coexistent : 30 ribosomes avec plus de 100 facteurs protéiques, 30 aminoacyl-ARNt synthétases, 340 ARN de transfert, 2 à 3 ARN messagers, 6 ARN polymérases, 330 protéines (dont 130 enzymes de la glycolyse et 100 enzymes du cycle de Krebs), 300 autres molécules, 30.000 petites molécules (H20, cofacteurs, précurseurs et métabolites) et 50.000 ions. Chez les Eucaryotes, le milieu intracellulaire contient en plus l'ensemble des protéines qui constituent le cytosquelette. Dans une expérience in vitro, ce même cube ne contient qu'une molécule d'enzyme, les sels du tampon et les molécules d'eau. Les effets de la sur-population intracellulaire sont non linéaires : plus la macromolécule a une masse (donc bien souvent une taille) importante, plus elle en subit les effets. Ces effets sont donc nettement moins importants pour les petits métabolites et les ions. Aucune espèce macromoléculaire n'est présente à forte concentration individuellement. Mais, ensemble, les macromolécules occupent une fraction importante du volume total : de 20 à 30 %. Cette fraction du volume intracellulaire n'est donc physiquement pas disponible pour d'autres molécules. Cette exclusion stérique a des conséquences considérables du point de vue énergétique :
|
Effets de la sur-population intracellulaire sur l'activité enzymatique Le milieu intracellulaire diffère donc considérablement d'une solution homogène (conditions in vitro) dans laquelle les macromolécules biologiques sont dispersées (très diluées) : les concentrations des enzymes in vivo sont de l'ordre de 10-5 à 10-4 M, valeurs très supérieures aux concentrations des enzymes in vitro (de l'ordre de 10-10 à 10-7 M). Une étude du métabolome de Escherichia coli a montré que la concentration des métabolites est supérieure à la valeur de leur KM pour la grande majorité des enzymes. |
Exemples d'effets de la sur-population intracellulaire sur les paramètres cinétiques de la catalyse par quelques enzymes La sur-population intracellulaire est simulée in vitro par l'addition, dans le milieu de mesure de l'activité enzymatique, d'agents tels que le polyéthylène glycol (PEG), le Ficoll, le dextran ou l'albumine de sérum bovin (BSA). |
||
L'activité enzymatique augmente puis diminue avec l'augmentation de la concentration en protéines. |
L'addition de PEG 4000 à 20.000 :
|
|
| L'activité spécifique de l'uréase augmente jusqu'à 10 fois en présence d'une concentration d'hémoglobine de 30% (en poids). | ||
| Le PEG 6000 augmente l'activité de l'enzyme AspP de Escherichia coli : une concentration de PEG 50 mg/mL diminue KM d'un facteur 4 et augmente VMax d'un facteur 6. | ||
| Le dextran 70.000, le Ficoll 70.000 ou le PEG 6000 augmente l'activité enzymatique de l'isochorismate synthase : une concentration de 25% de l'un de ces additifs, diminue KM d'un facteur 2 - 3. | ||
L'activité spécifique de l'hexokinase diminue à des concentrations élevées de BSA : une concentration de BSA de 250 mg/mL diminue kcat de 33 % et diminue KM de 25 %. Les effets des petits osmolytes et des concentrations élevées de BSA sur l'activité de l'hexokinase sont additifs. |
||
|
Voir un cours sur les protéases. a. Généralités Les protéines ou les peptides sont hydrolysées en fragments peptidiques par des protéases. Il en existe plusieurs familles constituées d'un trés grand nombre d'enzymes : |
| protéases à sérine | métallo-protéases | aspartyl-protéases | protéases à cystéine | protéases à thréonine |
et bien d'autres ... |
et bien d'autres ... |
et bien d'autres ...
|
et bien d'autres ... |
et bien d'autres ... |
Toutes les informations fonctionnelles (spécificité des substrats, inhibiteurs, séquences et autres) et structurales sont recensées dans des bases de données telles que : |
|
Les deux familles de protéases à sérine ("chymotrypsin-like" et "subtilisin-like") contiennent entre autres : la trypsine, la chymotrypsine, l'élastase, la subtilisine (voir tableau ci-dessus). Chymotrypsine : en 1901, H. M. Vernon a proposé que les préparations de pancréas contiennent un activateur intrinsèque à ses propres enzymes. Cette idée ne fût acceptée qu'en 1934 quand Moses Kunitz et John Howard Northrop ont confirmé la présence d'une enzyme en plus de la trypsine : ils l'ont nommée chymotrypsine. Ils l'ont cristallisée de même que son précurseur inactif, le chymotrypsinogène. En 1938, M. Kunitz a isolé différentes formes actives de la chymotrypsine en les appelant alpha, bêta et gamma. En 1947, Jacobsen a identifié des formes supplémentaires de la chymotrypsine en les appelant delta et pi. Le domaine protéique appelé Kunitz (ou domaine inhibiteur de protéases de type Kunitz) est un domaine qui inhibe les protéases. Il est relativement petit (50 à 60 acides aminés). Exemple : l'aprotinine ou inhibiteur de la trypsine pancréatique bovine (BPTI). En 1954, la première preuve du mécanisme catalytique en 3 étapes de l'hydrolyse de substrats amide et ester par la chymotrypsine a été apportée par Brian Hartley et B. Kilby. Ils ont émis l'hypothèse d'une forme acyl-enzyme intermédiaire, ce qui fut démontré ultèrieurement par Henderson en 1970. La chymotrypsine suit un mécanisme général de type "ping-pong". En 1955, Michael Laskowski Jr. a obtenu une deuxième forme cristalline du chymotrypsinogène (chymotrypsinogène B). Trypsine : en 1876 - 1877, Wilhelm Kühne a découvert la trypsine dans le liquide pancréatique de l'intestin de boeuf au cours de ses études sur la digestion intermédiaire des protéines dans le tractus gastro-intestinal. Il a conclu que la trypsine était initialement inactive puis convertie en sa forme active. Cette observation est à la base de la notion de précurseur inactif des protéases que l'on appelle zymogène et de l'activation de ce zymogène par (auto)protéolyse. En 1931, J.H. Northrop et M. Kunitz ont cristallisé la trypsine après la première purification de la pepsine en 1930. En 1974 la structure tridimensionnelle de la trypsine a été déterminée et a servi de prototype pour la famille des endopeptidases à sérine de type S1 à laquelle la trypsine appartient. De 1987 à 1992, des études de mutagénèse dirigée (trypsine recombinante) ont permis de déterminer le rôle d'acides aminés particulier. La trypsine est utilisée dans les protocoles de cultures de cellules et de tissus, dans l'identification des protéines par séquençage de peptides (protéomique et spectromètrie de masse). La trypsine est une protéase très étudiée dans divers domaines médicaux : la mutation R117H empêche son autolyse et provoque la pancréatite. La trypsine a été utilisée pour modéliser la décomposition du cartilage articulaire dans l'ostéo-arthrite. Elastase : on attribue à Christiaan Eijkman les premières études de l'élastase en 1904, comme produit des bactéries. Il a fallu attendre les études de J. Balo et I. Banga en 1949 pour démontrer qu'elle est une enzyme protéolytique du pancréas comme la trypsine et la chymotrypsine. En 1968, David Shotton et Brian Hartley ont obtenu les premiers cristaux d'élastase porcine. Le rôle de l'élastase dans l'emphysème, l'athérosclérose et la pancréatite aiguë hémorragique a été étudié entre 1968 et 1974. En 1974, W. Ardelt a découvert un deuxième type d'élastase appelée élastase pancréatique II. Les protéases à sérine sont probablement les enzymes les plus étudiées tant du point de vue du mécanisme catalytique (voir ci-dessous) avec un trés grand nombre d'inhibiteurs de toutes sortes, que du point de vue structural. Par exemple, on dénombre plus de 900 structures cristallographiques liées à la trypsine (trypsine, trypsinogène, Kunitz trypsin inhibitor, ...). Les protéases à sérine :
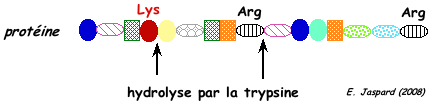
Par exemple, la trypsine hydrolyse la liaison peptidique après (côté C-terminal) les acides aminés lysine et arginine sauf si ces acides aminés sont suivis par une proline.
Cependant les protéases à sérine peuvent avoir une spécificité très complexe.
|
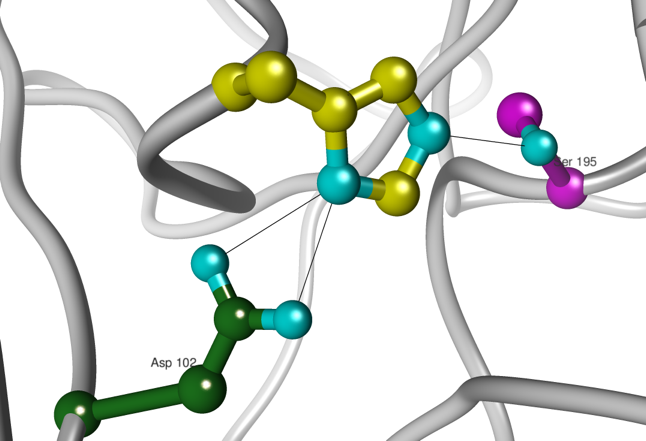
c. Les acides aminés du site catalytique des protéases à sérine : la "triade" catalytique Les protéases à sérine possèdent une sérine 195 réactive dans leur site actif. Deux autres acides aminés sont impliqués dans la catalyse par les protéases à sérine : l'histidine 57 et l'aspartate 102. Les protéases à sérine utilisent les deux types de catalyse :
Figure ci-dessous : les 3 acides aminés His 57, Asp 102 et Ser 195 forment la triade catalytique.
Source : E. Jaspard (2011) |
|
Visualisation de la triade catalytique dans la forme acyl-enzyme de l'élastase pancréatique à une résolution de 0,95 Å. Code PDB : 1GVK
|
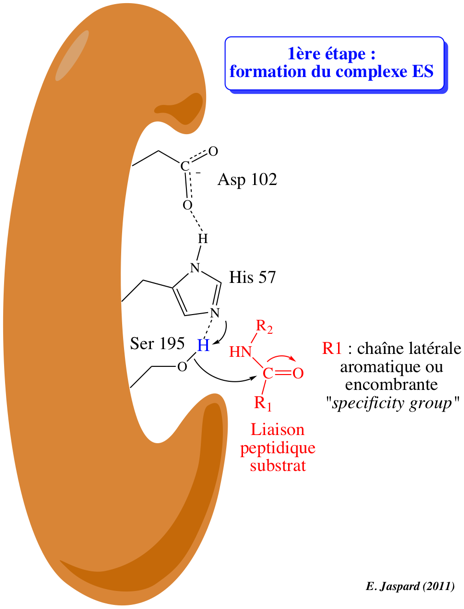
7. Mécanisme catalytique des protéases à sérine Voir un trés bon cours : Enzyme Mechanisms 1ere étape : formation du complexe enzyme-substrat (ES)
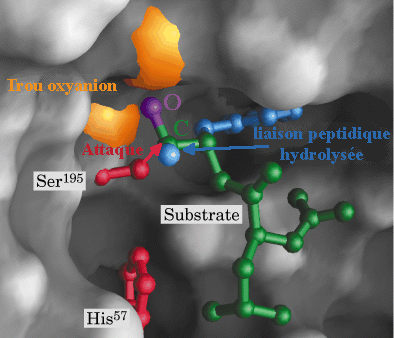
La protéase fixe le substrat (protéine ou peptide) dont elle hydrolyse la liaison peptidique au niveau d'acides aminés spécifiques de la protéase considérée. La chaîne latérale R1 de l'acide aminé impliqué dans l'hydrolyse de la liaison peptidique est de nature aromatique ou encombrante (dans le cas de la trypsine) mais de toute autre nature pour d'autres types de protéases. Cette chaîne latérale se fixe dans un endroit particulier du site actif de la protéase appelée poche de spécificité ("specificity pocket") de caractère hydrophobe. Cette fixation optimise l'orientation stéréochimique de la liaison peptidique afin qu'elle soit hydrolysée du côté C-terminal (carboxyle) du résidu d'acide aminé. Le squelette carboné du polypeptide substrat forme un court feuillet β avec le squelette carboné de l'enzyme au niveau du site actif (non montré dans la figure). L'oxygène du groupement carbonyle du substrat forme une liaison hydrogène avec un groupement NH (amide) du squelette carboné de l'enzyme (non montré dans la figure). Le complexe enzyme-substrat (ES) est ainsi formé. |
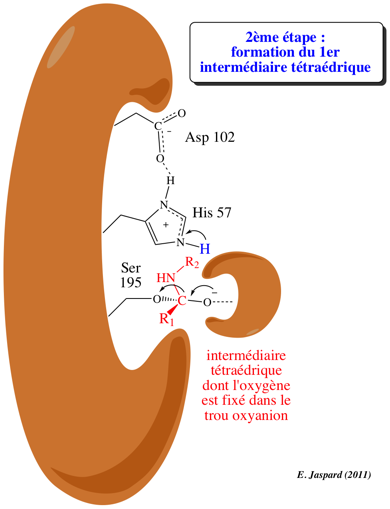
2ème étape : acylation / formation du premier intermédiaire tétraédrique
L'oxygène du groupement OH de la Ser 195 du site actif est activé en formant une liaison hydrogène avec l'azote N: du noyau imidazole de l'His 57. His 57 agit comme un accepteur de proton de Ser 195, selon un mécanisme de catalyse base générale, et devient His-H+. En même temps, l'oxygène Ser-O devient nucléophile et attaque le C du groupement carbonyle de la liaison petidique à hydrolyser. Une liaison covalente est ainsi formée selon le mécanisme de catalyse par covalence. Asp 102 a pour rôle le maintien d'un arrangement stéréospécifique idéal du réseau de liaisons hydrogène établies entre His 57 et Ser 195. Il stabilise aussi la forme His-H+ par interaction électrostatique, ce qui facilite le transfert du proton. Le résultat de cette étape dite d'acylation est la formation du premier intermédiaire tétraédrique dont la structure est probablement semblable à celle de l'état de transition (ES#) avec un oxygène carbonyle négatif appelé l'oxyanion. Le site actif fixe plus fortement l'oxyanion qu'il ne fixe le groupement carbonyle original du substrat : l'état de transition est ainsi stabilisé. Une autre liaison hydrogène est établie entre l'oxyanion et un groupement NH du squelette carboné de l'enzyme (au niveau de Gly 193) situé dans une région appelée "trou oxyanion". |
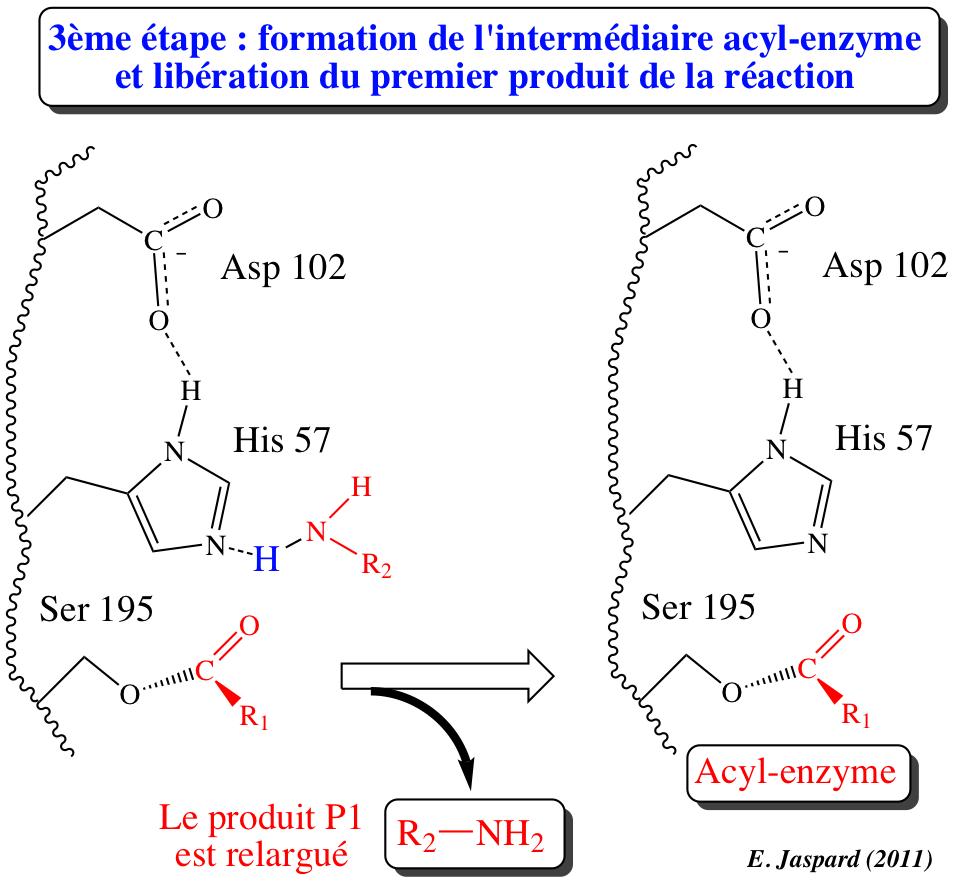
3ème étape : acylation (formation de l'acyl-enzyme) / libération du produit P1
La liaison peptidique originelle est clivée : His-H+ cède un proton à la moitié "amino" du substrat d'origine et agit selon un mécanisme de catalyse acide générale. Cette partie aminée est dissociée du site actif et le produit P1 (R2-NH2) est libéré. L'oxyanion redevient C=O qui reste attaché de manière covalente à Ser 195. C'est l'intermédiaire acyl-enzyme fixé par une liaison ester entre la moitié "carbonyle" du substrat d'origine et l'oxygène de la fonction alcool de Ser 195. |
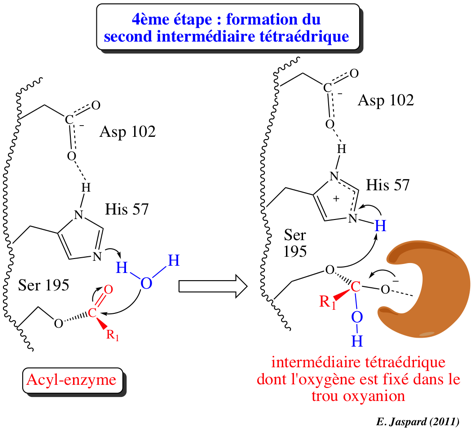
4ème étape : désacylation / formation du second intermédiaire tétraédrique
Le second substrat, la molécule d'eau, forme une liaison hydrogène avec His-N: et His 57 agit de nouveau selon un mécanisme de catalyse base générale, et devient His-H+. L'oxygène de l'eau est activé ce qui le rend nucléophile : il attaque le C du groupe carbonyle de l'acyl-enzyme (catalyse par covalence). Asp 102 a pour rôle le maintien d'un arrangement stéréospécifique idéal du réseau de liaisons hydrogène établies entre His 57 et Ser 195. Il stabilise aussi la forme His-H+ par interaction électrostatique ce qui facilite le transfert du proton. Le résultat de cette étape est la formation du second intermédiaire tétraédrique (groupement -OH au lieu d'un groupement amide dans le cas du premier) dont la structure est probablement semblable à celle de l'état de transition (ES#) avec un oxygène carbonyle négatif (oxyanion). Le site actif fixe plus fortement l'oxyanion qu'il ne fixe le groupement carbonyle original du substrat : l'état de transition est ainsi stabilisé. De nouveau, une autre liaison hydrogène est établie entre l'oxyanion et un groupement NH de Gly 193 situé dans le "trou oxyanion". |
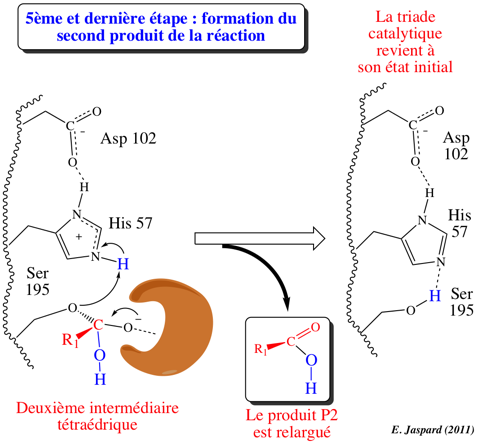
5ème étape : désacylation / libération de P2 / reformation de E
His-H+ redonne son proton à l'oxygène de la fonction alcool de Ser 195 de l'acyl-enzyme. La liaison ester est clivée, libérant le produit P2 : R1-COOH qui correspond à la partie N-terminale du substrat d'origine. Les 3 acides aminés de la triade catalytique retrouvent leur état initial. L'enzyme est de nouveau sous la forme E : elle est prête à catalyser de nouveau la réaction. |
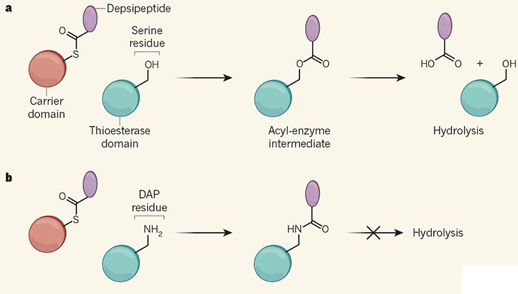
Stabilisation des intermédiaires d'une réaction enzymatique Les enzymes appelées peptides synthétases non ribosomales assemblent des peptides ou des depsipeptides (peptides contenant une ou plusieurs liaisons ester). Ces enzymes utilisent des domaines porteurs ("carrier domains") pour acheminer le produit de la réaction en cours de formation vers les domaines catalytiques pour l'extension. Le produit est enfin libéré du dernier domaine porteur par une réaction catalysée au sein du domaine thioestérase de la synthétase de la valinomycine. Un résidu d'acide aminé nucléophile (souvent, un résidu de sérine) du domaine thioestérase réagit avec la liaison qui attache le depsipeptide au domaine porteur : un intermédiaire acyl-enzyme est ainsi formé. Il est ensuite hydrolysé pour libérer le depsipeptide. Un procédé a récemment été mis au point (Huguenin-Dezot et al., 2018) au cours duquel le résidu nucléophile est remplacé par le résidu d'acide 2,3-diaminopropionique (DAP) : le groupe hydroxyle (OH) de la sérine est remplacé par un groupe amine (NH2). L'acyl-thioestérase intermédiaire qui en résulte résiste à l'hydrolyse : le premier et le dernier intermédiaire acyl-thioestérase ont ainsi pu être stabilisés sous forme de conjugué DAP et analysés par par cristallographie aux rayons X.
DAP : acide 2,3-diaminopropionique / Source : Gulick A.M. (2018) |
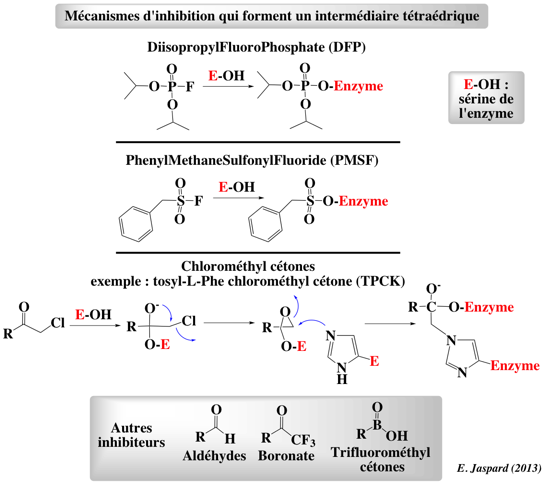
Figure ci-dessous : mécanismes d'inhibition des protéases à sérine par différents composés qui forment un intermédiaire tétraédrique.
Ces composés ont permis d'identifier la sérine 195 du site actif des protéases à sérine et d'en déterminer le rôle. |
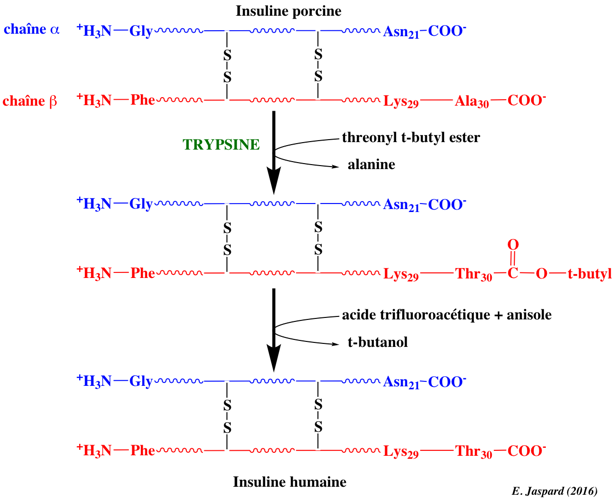
Illustration de la capacité de certines protéases à catalyser la synthèse peptidique (réaction réverse de l'hydrolyse peptidique). Exemple de la conversion de la séquence de l'insuline porcine en insuline humaine qui nécessite le remplacement de l'alanine 30 C-terminale de la chaîne β par une thréonine.
C'est réalisé par une étape de trans-peptidation catalysée par la trypsine en utilisant une threonine dont le groupement carboxyle est protégé en solution aqueuse avec un co-solvant organique. Le groupe protecteur est éliminé par hydrolyse douce. Voir un développement sur la synthèse peptidique par les protéases. |
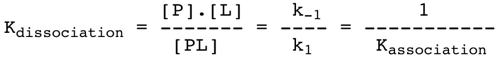
{kind=link}
{kind=link}