| Les glucides |
| Tweet |
|
|
I. DÉFINITION & RÔLES
II. STRUCTURE DES OSES A. Structure linéaireIII. PROPRIÉTÉS CHIMIQUES DES OSES A. Propriétés liées au groupement réducteurIV. ÉTUDE DE QUELQUES OSES ET DÉRIVÉS A. Oses simples |
V. ÉTUDE DE QUELQUES OSIDES ET DÉRIVÉS A. Définitions VI. Liens Internet et références bibliographiques |
|
Les glucides constituent un ensemble de substances dont les unités de base sont les sucres simples appelés oses ou monosaccharides. Les oses ont été définis comme des aldéhydes ou des cétones polyhydroxylées. Ce sont des composés hydrosolubles et réducteurs. Les glucides sont présents partout dans la biosphère et représentent en poids la classe prépondérante parmi les molécules organiques. La plus grande part des glucides amassés provient de la photosynthèse, processus qui incorpore le CO2 dans les glucides. Les glucides jouent plusieurs rôles capitaux dans les cellules :
Les glucides sous forme polymérisée sont appelés des osides. Ils peuvent être composés :
|
|
II. Structure
A. Structure linéaire
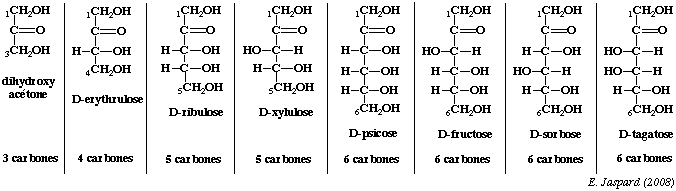
Les premiers oses qui ont un rôle sont des oses en C3 ou triose, il s'agit du glycéraldéhyde et la dihydroxyacétone. Il faut noter que sous leur forme phosphorylée ces deux composés correspondent à une étape importante de la voie de la glycolyse, puisqu'il s'agit du passage d'un sucre en C6 (le fructose 1, 6 diphosphate) à 2 sucres en C3. |
|
Le glycéraldéhyde possède un carbone dont les quatre substituants sont des groupes différents, il s'agit donc d'un carbone asymétrique ou chiral. Le glycéraldéhyde peut donc exister sous deux formes différentes (image l'une de l'autre dans un miroir et donc non superposables) qui correspondent à des configurations opposées autour du carbone chiral: les 2 composés sont appelées énantiomères. En 1906, Emil FISCHER et ROSANOFF ont choisi le glycéraldéhyde comme composé de référence pour l'étude de la configuration des sucres. Emil Fischer a choisi arbitrairement (ne disposant pas des méthodes physiques nécessaires à la détermination de la configuration absolue des oses) le symbole D pour l'énantiomère dextrogyre, c'est-à-dire le composé qui dévie le plan de la lumière polarisée vers la droite (dans le sens des aiguilles d'une montre). Ce n'est qu'en 1951 - 1954 que J.M. Bijvoet a montré par des études cristallographiques que le choix arbitraire de Fischer correspond à la configuration absolue des oses. Tous les oses dérivant du glycéraldéhyde dextrogyre ont été dits appartenir à la série D et tous ceux provenant du glycéraldéhyde lévogyre ont été dits appartenir à la série L. Configuration absolue : voir les règles de priorité de R.S. Cahn, C.K. Ingold et V. Prelog (1956). Stéréoisomère R ("rectus") : ordre de priorité dans le sens des aiguilles et stéréoisomère S ("sinister") : ordre de priorité dans le sens opposé. |
|
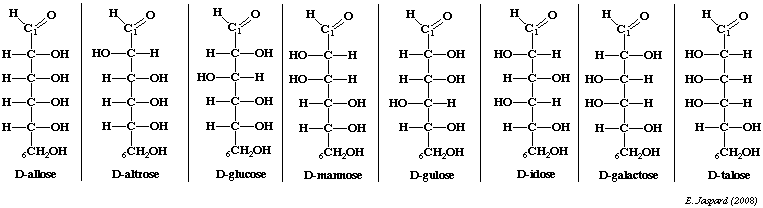
Tous les aldoses peuvent être synthétisés à partir du glycéraldéhyde. Dans la projection de Fischer, tous les oses dont l'hydroxyle porté par l'avant dernier carbone est à droite sont de la série D. Par ailleurs, dans cette projection, par convention, les liaisons représentées horizontalement pointent en avant du plan et les liaisons représentées verticalement pointent en arrière du plan. Quand on passe d'un ose à l'ose supérieur, un groupe H-C-OH chiral est ajouté entre le carbone terminal qui porte la fonction alcool primaire et le carbone carbonyle adjacent. A chaque addition, il existe 2 possibilités :
On peut citer l'exemple du glucose : c'est un ose à 6 carbones ou hexose. Il existe donc 16 stéréoisomères, 8 de la série D et 8 de la série L. Les sucres naturels sont en grande majorité de la série D.
Exemple : le D-mannose et le D-galactose sont des épimères du D-glucose mais ne le sont pas entre eux.
Série ascendante des D-cétoses et épimères de cétohexoses.
|
|
B. Structure cyclique 1. Hémiacétal et mutarotation. La structure linéaire ou structure à chaîne ouverte des oses ne rend pas compte de toutes leurs propriétés dés que le nombre des atomes est supérieur à 4. En premier lieu les propriétés réductrices qui ne sont pas tout à fait celles des aldéhydes et des cétones :
Ce phénomène a été appelé mutarotation par Lowry (1889). Rappel : Le pouvoir rotatoire spécifique [α]20D est mesuré avec un appareil qui s'appelle un polarimètre. On le définit en précisant la température, la longueur d'onde à laquelle est effectuée la mesure (il s'agit en général de la raie D du sodium 589 nm). Par ailleurs, la concentration est exprimée en g/ml et la longueur du tube du polarimètre est exprimée en décimètre. Connaissant le pouvoir rotatoire spécifique d'un composé, la loi de BIOT permet de déterminer la concentration d'une solution de ce même composé. Cette loi est additive, c'est-à-dire que le pouvoir rotatoire d'un mélange est la somme des pouvoirs rotatoires des composés qui constituent ce mélange. |
|
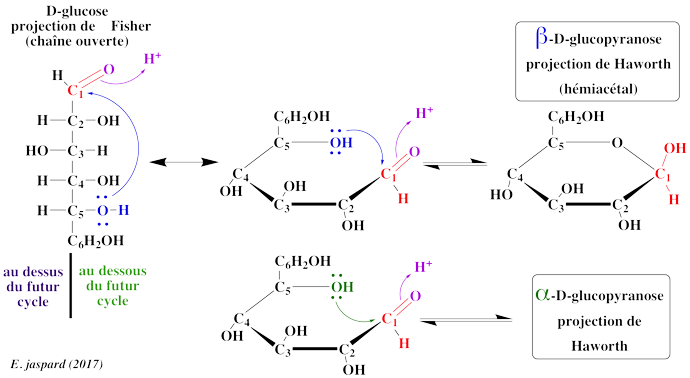
2. Mécanisme de cyclisation et représentation de Walter Norman Haworth. Le phénomène de mutarotation implique l'existence d'un carbone asymétrique supplémentaire. Par ailleurs, la formation d'hémiacétal implique que la fonction réductrice a déjà établit une liaison avec un alcool. C'est en 1884 que Bernhard Tollens a fourni l'explication par la structure cyclique des oses :
On obtient donc un nouveau carbone asymétrique et les deux isomères ne diffèrent que par la position d'un groupement sont appelés anomères. Le groupement hydroxyle porté par le carbone 4 peut également réagir et on obtient un cycle à 5 sommets ou cycle furanose. Les noms de pyranose et de furanose ont été adoptés par analogie avec les hydrocarbures à 6 et 5 sommets, respectivement. |
|
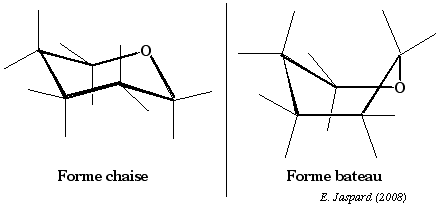
Les études de la stabilité conformationnelle du cyclohexane ont montré que les arrangements spatiaux qui ne subissent pas de contraintes stériques sont la conformation dite en chaise et d'autres, quelques peut moins stables, dont la principale est la conformation dite bateau.
La position des substituants hydrogène peut être soit dans un axe perpendiculaire aux plan défini par les 6 liaisons carbone-carbone, ce sont des substituants dits axiaux, soit au contraire dirigés vers l'extérieur de ce cycle et ils sont dits équatoriaux. Dans le cas du glucopyranose, c'est essentiellement la forme chaise qui existe. |
|
III. Propriétés chimiques A. Propriétés liées au groupement réducteur Les oses sont des réducteurs plus faibles que les aldéhydes ou les cétones vrais. Le résultat de l'oxydation dépend des conditions de cette oxydation. a) Par oxydation douce des aldoses avec Br2 ou I2 en milieu alcalin, on obtient les acides aldoniques :
b) Par oxydation plus poussée avec l'acide nitrique à chaud on obtient les acides aldariques qui sont des diacides possédant une fonction carboxylique sur le carbone 1 et le carbone 6:
c) Enfin, si la fonction aldéhyde est protégée pendant l'oxydation, on obtient les acides uroniques oxydés uniquement sur la fonction alcool primaire :
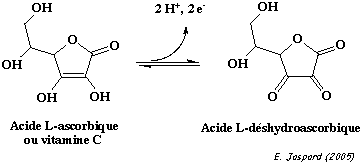
Ces composés interviennent dans la reconnaissance cellulaire chez les bactéries. L'acide glucuronique est le précurseur de la voie de synthèse de la vitamine C ou acide L-ascorbique. La vitamine C est l'ènediol d'une lactone d'acide aldonique. Le pKa du groupe hydroxyle en C3 de ce dérivé glucidique est relativement bas du fait de la stabilisation par résonance de sa base conjuguée. |
|
Les réactions de réduction se font par hydrogénation catalytique, soit par action d'un borohydrure alcalin tel que LiBH4 ou NaBH4.
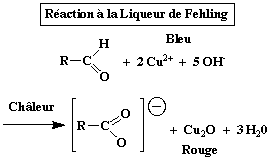
Il faut mentionner d'autres réactions de réduction utilisées pour le dosage des sucres et leur caractérisation. Notamment :
|
|
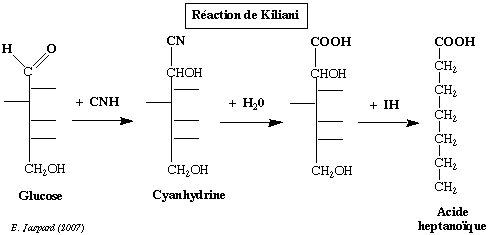
a) avec le cyanure et l'hydroxylamine Les réactions de condensation incluent la synthèse de KILIANI et la dégradation de WOHL - ZEMPLEN. Ces deux voies permettent de passer d'un ose à respectivement l'ose supérieur et l'ose inférieur. Elles ont toutes deux permis d'établir la filiation des oses avec le glycéraldéhyde. La synthèse de KILIANI : le glucose réagit avec l'acide cyanhidrique pour former une cyanhidrine (2 stéréoisomères) qui, après hydrolyse, donne un acide hexahydroxylé.
Celui-ci est réduit par IH (en présence de phosphore rouge) et donne l'acide heptanoïque. La même réaction à partir du fructose donne l'acide méthyl 2-hexanoïque. b) avec les alcools et les phénols Cette réaction est tout particulièrement importante. En effet, les substances obtenues sont les osides ou glycosides et la liaison qui joint l'ose à l'alcool ou au phénol est la liaison O-osidique ou glycosidique. Il est important de noter que la formation de cette liaison s'accompagne de la perte du pouvoir réducteur de l'ose et blocage de la configuration du cycle. |
|
B. Propriétés liées aux fonctions alcooliques Ils permettent d'effectuer des éléctrophorèses des oses, ce qui n'est pas possible sans celà puisque les oses ne sont pas chargés naturellement. De plus ils ont permis de démontrer que dans l'α-D-glucose, l'hydroxyle du carbone anomère est en position cis par rapport à l'hydroxyle porté par le carbone 2, donc qu'il se situe en dessous du cycle. En effet, le complexe se forme plus aisément avec l'anomère cis qu'avec l'anomère trans. L'anomérie du sucre influencera la formation des complexes avec le bore et donc leur mobilité éléctrophorétique. |
|
Les agents méthylants tels que le sulfate de méthyle ((SO4(CH3)2) en présence de soude (Haworth) ou l'iodure de méthyle ICH3 avec Ag2O (Purdie) agissent en substituant tous les hydrogènes des groupements hydroxyles par un -CH3 formant ainsi un groupement éther. Si le groupement réducteur de l'ose est libre, il réagira en formant un dérivé O-méthylé.
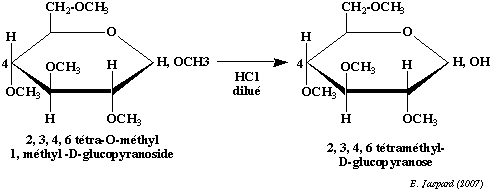
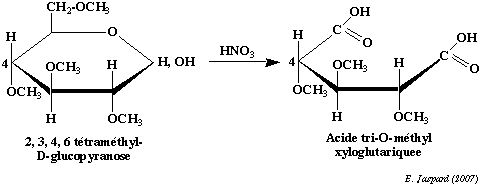
Cependant, cette liaison est une liaison osidique qui n'a pas la même stabilité en milieu acide où elle est facilement hydrolysée. Il faudra donc la distinguer des liaisons ether en la spécifiant dans la nomenclature de l'ose. La méthylation est une technique importante qui a deux applications principales : a) en premier lieu elle permet de déterminer la structure des cycles :
On méthyle complètement un ose cyclique, puis on hydrolyse la liaison osidique en milieu acide dilué. On oxyde ensuite le composé par l'acide nitrique. L'oxydation rompt le cycle et élimine les carbones qui ne font pas partie du cycle, en l'occurence le carbone 6 dans le cas d'un pyranose et les carbones 5 et 6 dans le cas d'un furanose. Le reste du cycle se retrouve sous la forme d'un diacide tri-O-méthylé dans le cas d'un pyranose et d'un diacide di-O-méthylé dans le cas d'un furanose. b) en second lieu elle permet de déterminer l'enchaînement dans les polyosides : On méthyle complètement un oside et on coupe ensuite les liaisons osidiques en milieu acide dilué. On peut voir dans l'exemple de l'amylose que le composé terminal non réducteur (donc le composé dont l'hydroxyle hémiacétalique est impliqué dans la liaison osidique mais, inversement, dont le carbone 4 n'est pas impliqué dans cette liaison) donnera un dérivé tétra-O-méthylé alors que tous les autres éléments donneront un dérivé tri-O-méthylé. Dans le cas d'une structure branchée, on obtiendra des dérivés di-O-méthylés pour chaque ose impliqué dans le branchement (en l'occurence, l'ose qui a son carbone 6 impliqué dans la liaison osidique). |
|
IV. Étude de quelques oses et dérivés 1. Le glucose (aldohexose - pyranose) Le glucose est extrèmement répandu dans le règne végétal et le règne animal à l'état libre ou combiné à d'autres oses, sous forme phosphorylé ou non. C'est le combustible de la cellule, mis en réserve sous forme de glycogène (règne animal) ou d'amidon (règne végétal). Le métabolisme du glucose correspond à la voie de la glycolyse et aux voies qui en découlent. D'un point de vue structural, en ce qui concerne les oses, il faut faire attention à la position de l'hydroxyle de l'avant dernier carbone (le C5 pour un hexose) dans la projection de Fischer, et la position du C6 qui en découle dans la représentation de Haworth. En effet, dans la représentation de Haworth, c'est au niveau du C6 (pour un hexose) que l'on peut savoir s'il s'agit de la série D ou L puisque l'hydroxyle du C5 est engagé dans l'hémiacétal et n'indique donc plus la série de l'ose. Si on représente l'ose comme en (1), on ne précise pas la nomenclature du carbone anomère (qui peut donc être α ou β). |
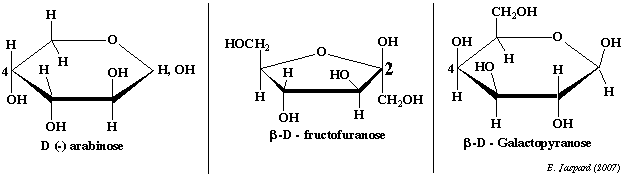
2. L'arabinose (aldopentose - pyranose) L'arabinose (figure ci-dessous) est abondant dans le monde végétal. Il contribue à la formation des tissus de soutien. D'un point de vue structural, en ce qui concerne les oses, il en va de même pour un pentose comme l'arabinose mais dans ce cas il faut regarder au niveau du C4. 3. Le fructose (cétohexose - furanose) C'est le cétose que l'on obtient par interconversion du glucose et du mannose, c'est à dire par épimérisation du C2 du glucose. Le fructose (figure ci-dessous) est un ose qui souligne l'importance de la forme linéaire : en effet, en ce qui concerne cet ose, la forme linéaire est toujours présente à concentration élévée et on a aussi un équilibre avec les formes furaniques qui sont les formes les plus stables à l'état naturel. Voir entrée d'autres oses que le glucose dans la glycolyse. 4. Le galactose et le mannose (aldohexose - pyranose) Ces deux oses sont beaucoup moins abondant dans les cellules que le glucose mais on les trouve comme constituants des glycoprotéines et des glycolipides.
|
|
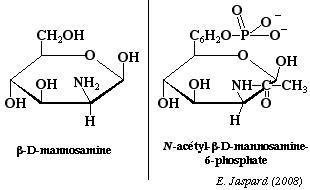
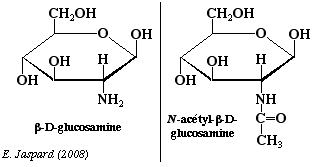
Les osamines sont synthétisées à partir du fructose-6-phosphate et sont obtenues par substitution de l'hydroxyle du carbone 2 par un NH2. Le groupement aminé est le plus souvent acétylé. Ce sont des oses trés importants: par exemple, la chitine, polyoside constitutif de la carapace des insectes est un polymère de la N-acétylglucosamine avec des liaisons β-1,4. La N-acétyl-mannosamine-6-phosphate est le précurseur des acides sialiques. |
|
Ce sont des composés caractéristiques des glycoprotéines. Ils s'y trouvent en bout de chaîne liés par une liaison α-glycosidique. La fonction COOH est libre, ce qui confère aux glycoproyéines un caractère acide marqué. Les acides sialiques dérivent tous de l'acide neuraminique dont le plus courant est l'acide N-acétyl neuraminique.
La synthèse est obtenue à partir de La N-acétyl-mannosamine-6-phosphate et du phosphoénol pyruvate.
Dans les glycoprotéines, les acides sialiques sont disposés à intervalles réguliers le long de la chaîne. Ils forment ainsi un nuage électronégatif qui, par répulsion électrostatique, maintient la chaîne allongée sous forme de bâtonnet. Il s'ensuit une grande viscosité de ces composés. |
|
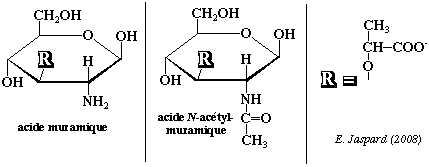
L'acide muramique N-acétylé est un composant de la muréine, haut polymère de nature glycopeptidique qui forme le support fondamental des parois bactériennes. L'acide muramique N-acétylé dérive lui-même de la N-acétyl-glucosamine. La biosynthèse se fait également à partir du phosphoénol pyruvate.
Il y a formation d'esters phosphoriques sous l'action de kinases qui transfèrent le groupe phosphate terminal de l'ATP. Utilisés comme source d'énergie, c'est sous leurs formes phosphorylées que les oses sont interconvertis et donc métabolisés (voie de la glycolyse et voie des pentoses phosphate par exemple). La liaison ester-phosphate est hydrolysée par des phosphatases. Les esters phosphoriques du glucose et du fructose peuvent être considérés comme les produits de l'assimilation photosynthétique. Le β-D-ribose-5-phosphate et le β-2-désoxy-D-ribose-5-phosphate sont par ailleurs les deux oses constitutifs des acides nucléiques. |
|
V. Étude de quelques osides et dérivés Les osides ou glycosides sont des substances dans lesquelles l'hydroxyle du groupement hémiacétalique du carbone anomèrique d'un ose a été condensé avec un groupement hydroxylique (alcoolique ou phénolique). La liaison qui joint l'ose à l'alcool ou au phénol est appelée 0-osidique ou glycosidique. Les osides donnent par hydrolyse au moins deux oses. B. Détermination de la structure d'oside a) détermination de la nature des oses constitutifs Les osides sont hydrolysés en milieu acide ou par voie enzymatique, de manière à rompre les liaisons osidiques. Dans le cas d'hétérosides, il faut déterminer la nature de l'aglycone. Puis ils sont séparés par des techniques chromatographiques et identifiés et dosés individuellement. b) détermination du mode de liaison entre les oses constitutifs On marque toutes les fonctions hydroxyles libres (par exemple, par méthylation et par l'acide périodique). Une hydrolyse acide différencie ensuite les liaisons éther-oxydes des liaisons osidiques. Dans le cas de polyosides complexes, il faut faire en plus des hydrolyses ménagées (incomplètes) menant à des oligosides (séparés par des techniques chromatographiques) dont l'étude complète cette détermination. Enfin, la détermination, s'il y a lieu, de l'anomérie de la liaison osidique fait appel à des enzymes spécifiques de chaque type de liaison ou, dans le cas le plus simple, par l'étude de la mutarotation après hydrolyse. c) détermination du caractère réducteur ou non de l'oside La technique de réduction par le borohydrure de Na permet de caractériser l'ose terminal réducteur dans le cas d'un dioside ou d'un oligoside. Dans le cas d'un polyoside, la proportion de l'ose réducteur terminal est si faible qu'il faut employer du borohydrure marqué par le tritium. d) détermination de la masse molaire et de la longueur des chaînes dans le cas des polyosides Les techniques physiques usuelles (osmométrie, ultracentrifugation, diffusion de la lumière, viscosimétrie, filtration sur tamis moléculaire, électrophorèse de complexes avec le bore...) auxquelles on peut joindre des techniques biochimiques (enzymes spécifiques de dégradation ou au contraire de synthèse in vitro) et immunochimiques. |
|
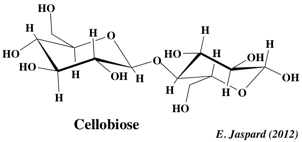
C. Quelques diholosides trés importants 1. Le maltose Ce diholoside est libéré par hydrolyse de l'amylose (voir ci-après), qui est un polymère de résidus glucose: il s'agit de l'α-D-glucopyranosyl-(1,4)-D-glucopyranose. Les résidus de glucose sont libérés par hydrolyse chimique ou par une enzyme: l'α-D-glucosidase. C'est un sucre réducteur puisque l'hydroxyle du carbone anomère du second glucose est libre.La méthylation suivie d'hydrolyse donnera donc du 2,3,6 tri-O-méthyl-glucose et du 2,3,4,6 tétra-O-méthyl-glucose. 2. Le lactose C'est le sucre du lait, propre au règne animal, synthétisé dans les glandes mammaires. Il s'agit du β-D-galactopyranosyl-(1,4)-D-glucopyranose. C'est le seul diholoside reducteur trouvé à l'état naturel. C'est un sucre extrémement représenté dans le règne végétal et tout particulièrement dans la canne à sucre et la betterave. Avec le tréhalose, c'est le seul diholoside non reducteur trouvé à l'état naturel (l'hydroxyle du carbone anomère du fructose est engagé dans la liaison osidique avec le glucose). Ainsi, on peut considérer le saccharose comme étant l'α-D-glucopyranosyl-β-D-fructofuranoside ou le β-D-fructofuranosyl-α-D-glucopyranoside. L'appellation de ce sucre explicite les 2 carbones impliqués (liaison 1,2 ou 2,1). La méthylation suivie d'hydrolyse donnera donc du 3,4,6 tri-O-méthyl-fructose et du 2,3,4,6 tétra-O-méthyl-glucose. Enfin, les solutions de saccharose présentent un pouvoir rotatoire mais pas le phénomène de mutarotation. Ce sucre provient de la dégradation de la cellulose. Il s'agit du β-D-glucopyranosyl-(1,4)-D-glucopyranose.
C'est donc un épimère du lactose (épimère en C4 du premier résidu de glucose). |
|
D. Deux triholosides: le gentianose et le raffinose Ces triholosides sont dérivés du saccharose :
Cités à titre d'exemple, notamment en ce qui concerne les thiohétérosides de synthèse (les thiogalactosides, par exemple) utilisés comme analogues de substrats ou inhibiteurs des réactions enzymatiques. |
|
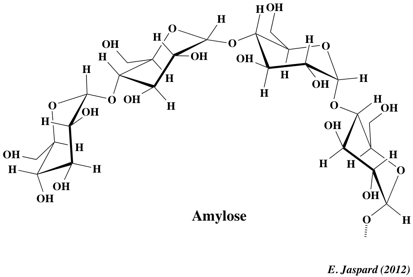
1. L'amidon C'est le polyoside de réserve des végétaux (exemples : pomme de terre, blé, maïs, banane ...). L'amidon des industries alimentaires ou non alimentaires est essentiellement produit à partir des céréales (exemples : maïs, blé ...). L'amidon est en fait un mélange de deux polysaccharides : l'amylose et l'amylopectine. Les proportions relatives d'amylose et d'amylopectine influencent les propriétés physiques de l'amidon. L'amidon est insoluble dans l'eau froide, et forme un empois (dispersion visqueuse) quand on chauffe le mélange. a. L'amylose : elle représente 15 à 30% de la masse de l'amidon. C'est un polymère linéaire de résidus glucose (500 à 20000 résidus - MM = 105 - 106 Da) liés par une liaison α-(1,4)-D-glucosidique. Les ramifications via une liaison α-(1,6)-D-glucosidique ne sont que d'environ 1%. Cette longue chaîne a une forme d'hélice simple gauche :
La chaîne est stabilisée par des liaisons hydrogène entre les groupements hydroxyle et les molécules d'eau.
Cette structure confère 2 propriétés à l'amylose :
|
b. L'amylopectine : elle représente 70 à 85% de la masse de l'amidon. Elle diffère de l'amylose du fait qu'il s'agit d'un polymère ramifié:
Plusieurs résultats ont permis de cerner l'arrangement de l'amylopectine:
Ces résultats et l'étude de certains modèles ont permis de montrer que l'on trouve en moyenne une ramification tous les 25 résidus et les branches contiennent une vingtaine de résidus. De plus les branchements sont plus ressérés du côté de l'extrémité réductrice de la chaîne. Enfin, certaines branches sont elles mêmes ramifiées. |
|
Visualisation de la β-amylase de Glycine max à une résolution de 1,90 Å. PDB : 1BYB La β-amylase (EC 3.2.1.2) hydrolyse la liaison α-(1,4)-D-glucosidique. |
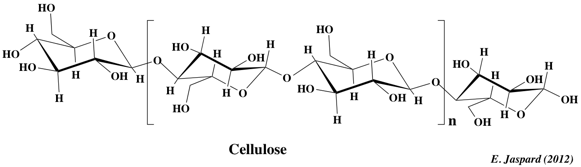
2. La cellulose La cellulose est d'origine végétale seulement. C'est une substance de soutien, puisqu'elle est le constituant principal de la paroi des cellules jeunes des végétaux. C'est la biomolécule la plus importante en masse à la surface de la terre et elle contiendrait la moitié du carbone disponible sur la terre. Elle est constituée de longues chaînes linéaires (300 à 1500 résidus - MM = 5 104 - 2,5 106 Da) de glucose lié en β-(1,4) D-glucosidique. L'unité de répétition est le cellobiose (voir plus haut).
La cellulose se caractérise par une grande inertie chimique. Bien qu'insoluble dans l'eau, elle a un caractère hydrophile : la fixation de très nombreuses molécules d'eau entraîne le gonflement de la cellulose, c'est un hydrocolloïde. La cellulose n'est pas attaquable par les sucs digestifs des omnivores: l'homme est incapable de digérer la cellulose car il est dépourvu d'enzymes actifs sur les liaisons β-glucosidiques. De plus, les enzymes qui la dégradent, les cellulases, sont trés peu répandues : les ruminants, les escargots et certaines bactéries. |
3. Applications industrielles Aller au site : "Polysaccharides alimentaires" - USTL L'industrie agroalimentaire utilise des polysaccharides aux propriétés stabilisantes et épaississantes (exemples : amidon, cellulose, hémicellulose, xanthane). 4. Le glycogène Le Le glycogène est le polyoside de réserve des animaux. Le stock principal se trouve dans le foie (200g pour un adulte) et dans les muscles (100 à 300 g). Le cerveau est un grand utilisateur de glucose : 100 mg/min, mais il ne possède qu'une réserve limitée de glycogène (10 à 20 g). Le glycogène ressemble beaucoup à l'amylopectine : il s'agit de chaîne de glucose liés en α-(1,4) et de branchements en α-(1,6). Cependant les chaînes sont beaucoup plus courtes et la molécule de glycogène est plus dense. Le glycogène est dégradé par des amylases comme l'amidon. Voir un cours sur la régulation de la glycogènolyse. 5. Les enzymes de dégradation des glucanes Les enzymes de dégradation des glucanes sont: a) Les amylases qui coupent spécifiquement les liaisons α-1,4: Les β-amylases à -SH actif sont trouvées dans le monde végétal. Elles coupent une liaison sur deux à partir de l'extrémité non réductrice, libérant ainsi des unités maltosyle. Les β-amylases ne coupent pas les liaisons α-1,6 et n'agissent donc que sur les chaînes externes des polysaccharides branchés. Les α-amylases sont des métalloenzymes et sont trouvées dans les deux règnes. Elles coupent les liaisons α-1,4 à l'intérieur des chaînes en formant des oligosides de petite taille (3 à 8 restes) qui peuvent contenir 1 ou 2 points de branchement puisqu'elles ne coupent pas les liaisons α-1,6. b) Les phosphorylases: elles attaquent les chaînes à partir des extrémités non réductrices, par phosphorolyse des liaisons α-1,4, avec libération d'α-D-glucose-1-phosphate. c) Les enzymes de débranchement: elles coupent les liaisons α-1,6 des points de branchement selon des modes différents en fonction de leur origine. |
|
1. Les mucopolysaccharides Ce sont des composés hétérogènes qui résultent de la condensation d'un nombre élévé de sous-unités disaccharidiques élémentaires. Cette unité est constituée :
Ce sont des molécules à caractère acide trés marqué. Elles sont toujours liées à une partie protéique. Cependant, dans le composé final, les glucides sont trés majoritaires (95%). Le plus simple des mucopolysaccharides est l'acide hyaluronique constitué de une molécule de N-acétyl-glucosamine β-(1,4) et d'une molécule d'acide glucuronique. Sa fonction principale, liée à la grande viscosité qu'il procure aux solutions, est de s'opposer à la diffusion de substances étrangères. 2. Les glycoprotéines Ces composés sont constitués d'une partie glucidique et d'une partie protéique. La partie glucidique varie , en poids, de 1 à 50% de la masse de l'ensemble. Les chaînes polysaccharidiques sont souvent ramifiées. Il existe des polysaccharides liés à O, comme le galactose lié au groupement hydroxyle d'une hydroxylysine dans le collagène. Cependant, les acides aminés impliqués sont souvent la sérine ou la thréonine. Les polysaccharides liés à N, sont unis par covalence à l'azote de la liaison peptidique de certaines asparagines. La glycosylation est un évènement post-traductionnel qui n'a lieu que chez les eucaryotes. Les protéines glycosylées sont destinées à être sécrétées ou à être intégrée à la membrane plasmique. La détermination de la structure des glycoprotéines est actuellement l'un des travaux les plus difficiles. La raison en est simple : chaque ose possède plusieurs hydroxyles libres et chacun peut établir une liaison avec un autre ose ou un autre composé. Ainsi, le nombre de polysaccharides qui peut être formé est immense. Par exemple, avec seulement trois oses, il existe plusieurs centaines de configurations. |
| VI. Liens Internet et références bibliographiques |
| "Principes de Biochimie" Horton, Moran, Ochs, Rawn et Scrimgeour (1994) - Ed. DeBoeck Universités - ISBN : 2-8041-1578-X | |
GlyCosmos : portail Web visant à intégrer les glycosciences aux sciences de la vie. Yamada et al. (2020) "The GlyCosmos Portal: a unified and comprehensive web resource for the glycosciences" Nat. Methods 17, 649 - 650 |
|
"Chapter 25: Carbohydrates" - University of Calgary "Polysaccharides alimentaires" (trés bon site pédagogique avec des quizz) |
|
| LEA Database | Aller au site |
![]()