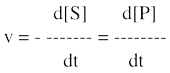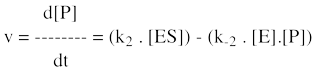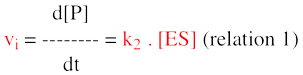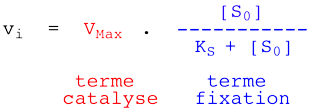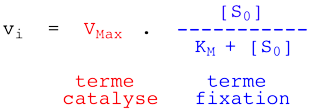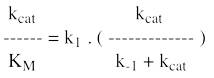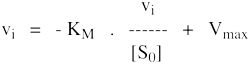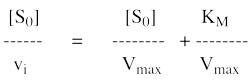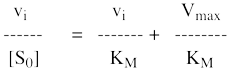| L'équation de Henri - Michaelis - Menten / L'équation de Briggs - Haldane |
| Tweet |
|
|
1. L'enregistrement de la cinétique de la réaction enzymatique 2. Vitesse d'apparition du produit 3. La mesure de la vitesse initiale de la réaction enzymatique 4. Conditions de concentrations en substrat pour les mesures de cinétiques enzymatiques : [S0] >> [E0] 5. L'hypothèse du quasi équilibre : l'équation de Henri, Michaelis et Menten (1913) |
6. L'hypothèse de l'état stationnaire de la concentration du complexe ES : l'équation de Briggs - Haldane (1925) 7. La signification des paramètres cinétiques : 8. Les représentations graphiques - Les méthodes modernes par calculs |
| 1. L'enregistrement
de la cinétique de la réaction enzymatique
L'enzymologie est une discipline qui paraît bien souvent théorique et, en effet, des modèles sont développés pour rendre compte de la sophistication et de la diversité des réactions enzymatiques. Cependant, les équations qui sont manipulées s'appliquent à l'analyse de données expérimentales : les valeurs des vitesses d'une réaction enzymatique mesurées dans diverses conditions (de concentrations de substrat, en présence de molécules qui augmentent ou diminuent la vitesse, à différents pH, ...). La cinétique d'une réaction enzymatique est la variation en fonction du temps de la concentration de molécules (substrats et produits de la réaction). L'enregistrement de cette cinétique implique que ces molécules possèdent des propriétés physico-chimiques qui permettent de les détecter de manière quantitative :
|
2. Vitesse globale d'apparition du produit Considérons le mécanisme catalytique le plus simple qui décrit :
La vitesse de la réaction est la variation de la concentration du substrat ([S]) ou celle du produit ([P]) en fonction du temps. Par convention, le signe moins indique que le substrat S disparaît.
Au début de la réaction enzymatique, il n'y a que le substrat et pas de produit. Il est donc préférable, du point de vue de la précision, de mesurer la vitesse d'apparition du produit plutot que la vitesse de disparition du substrat. En effet, on mesure plus précisément une faible augmentation de la concentration du produit (puisque l'on part de zéro), qu'une faible diminution de la concentration du substrat (qui est "énorme" au début de la réaction). L'équation de la vitesse globale liée à la concentration du produit ([P]) est la somme des vitesses de formation et de disparition du produit :
|
|
3. La mesure de la vitesse initiale de la réaction enzymatique Rappelons que Henri, Michaelis et Menten ont développé leur théorie à une époque (1913) où l'on ne disposait pas de moyens de calcul susceptibles d'approcher facilement les solutions de cette équation différentielle. Pour simplifier l'équation globale, il est nécessaire de faire une première approximation (tout à fait justifiée) : on considère qu'il existe un temps suffisamment court pendant lequel la quantité, donc la concentration, du produit formé est négligeable. Remarque : négligeable ne signifie pas nulle, sinon on n'aurait aucun signal détécté pour enregistrer la cinétique. Puisque le terme [P] est négligeable, le terme (k-2 . [E] [P]) l'est aussi. Remarque : celà ne signifie pas que la constante de vitesse k-2 est nulle ou négligeable (on ne sait rien à priori sur sa valeur). L'équation de la vitesse globale de formation du produit est simplifiée :
Du point de vue expérimental :
|
|
4. Conditions de concentrations en substrat pour les mesures de cinétiques enzymatiques : [S0] >> [E0] Une condition expérimentale* supplémentaire pour aboutir à l'équation de Henri, Michaelis et Menten est d'effectuer des mesures de cinétiques avec une concentration initiale du substrat [S0] beaucoup plus grande que la concentration initiale de l'enzyme [E0] et ce, pour toutes les concentrations en substrat étudiées. Avant la réaction enzymatique, le milieu réactionnel contient une concentration initiale connue de substrat [S0] et on déclenche la réaction en ajoutant l'enzyme à une concentration initiale connue [E0]. Dès l'addition de l'enzyme, le milieu réactionnel contient :
En ce qui concerne le substrat, la loi de conservation des espèces moléculaires s'écrit : [S0] = [S] + [ES] + [P]
Finalement, on peut considérer à tous moments de la réaction que : [S0] # [S] (relation 2) *Remarque : cette condition de concentration de substrat ne reflète pas la réalité cellulaire. Dans la cellule, les concentrations des molécules de substrats sont souvent très largement inférieures à celle des enzymes ([Scellule] << [Ecellule]). |
5. L'hypothèse du quasi équilibre : l'équation de Henri, Michaelis et Menten (1913) Articles originels :
Maud Menten (1879 - 1960)
Leonor Michaelis (1875 1949) Ces auteurs ont fait l'hypothèse que, dès l'addition de l'enzyme, il s'établit un équilibre rapide entre les formes libres de l'enzyme et du substrat (E et S) et le complexe enzyme-substrat (ES), appelé complexe de Michaelis - Menten :
On aboutit à la relation de Henri - Michaelis - Menten :
|
6. L'hypothèse de l'état stationnaire de la concentration du complexe ES : l'équation de Briggs - Haldane (1925) Ces auteurs ont développé une équation plus générale. Ils ont montré qu'il ne s'établit pas forcément un équilibre rapide pour toutes les enzymes entre les formes libres de l'enzyme et du substrat (E et S) et le complexe enzyme-substrat (ES).
Après un certain délai, appelé état pré-stationnaire, c'est la concentration du complexe ES qui est constante : cette concentration est dite à l'état stationnaire. Celà signifie que si [ES] = constante, les vitesses globales :
sont égales. Par ailleurs, si les mesures de cinétiques sont faites en condition de vitesse initiale, le terme (k-2.[E].[P]) est négligeable.
On aboutit à la relation de Briggs - Haldane :
|
Cas où [S] ne peut etre approximée à [S0] Lorsque KM est inférieur à la concentration en enzyme totale [E0], pour de faibles concentrations en substrat, la fraction du substrat liée à l'enzyme est non négligeable : l'approximation [S] = [S0] n'est plus valable. Donc si : [E] = [E0] - [ES] et [S] = [E0] => k1 . ([E0] - [ES]) . ([S0] - [ES]) = (k-1 + kcat) . [ES] => [ES]2 - ([E0] + [S0] + KM) . [ES] + ([E0] . [S0]) = 0 C'est une équation du second degré du type [ax2 + bx + c = 0 ], avec : a = 1; b = - ([E0] + [S0] + KM); c = ([E0] . [S0]) Dont la solution réelle est : vi = Vmax . {([E0] + [S0] + KM) - √([E0] + [S0] + KM)2 - 4 . ([E0] . [S0])} / 2 . ([E0] Morrison, J. F. (1969) "Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors" BBA - Enzymology 185, 269 - 286
Source : "The Quadratic Velocity Equation for Tight-Binding Substrates"
En règle générale, si la valeur de KM est inférieure à 5 [E0], l'équation du second degré de Morrison doit être utilisée de préférence à l'équation de Henri - Michaelis - Menten. |
|
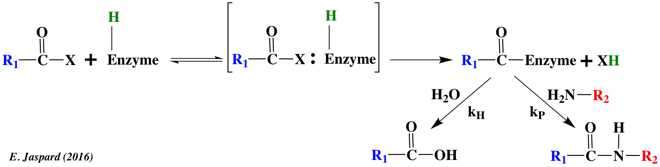
b. La constante de vitesse k-2 La constante de vitesse k-2 traduit la possibilité physique d'une même enzyme à catalyser la réaction réverse. Ce n'est pas systématiquement le cas. Exemples d'enzymes qui catalysent une réaction dans les deux sens 1er exemple : la phosphorylation (par l'ATP) du 3-phosphoglycérate en 1,3-bisphosphoglycérate est catalysée par la 3-phosphoglycérate kinase (EC 2.7.2.3) dans le cycle de Calvin. Cette enzyme catalyse la réaction réverse dans la glycolyse. 2ème exemple : la thermolysine (EC 3.4.24.27) est une metalloprotéase qui catalyse l'hydrolyse de la liaison peptidique impliquant des acides aminés hydrophobes : Leu / Ile / Phe / Trp / Tyr et Val. La coupure se fait avant ces acides aminés. Elle est aussi utilisée pour son aptitude à catalyser la réaction réverse : la formation de la liaison peptidique.
La synthèse peptidique est contrôlée cinétiquement si le taux de synthèse du peptide produit (kP) est élevé par rapport à la vitesse d'hydrolyse du peptide (kH). C'est le cas si l'on fournit un acide aminé ou un peptide qui soit un nucléophile plus puissant que l'eau pour accepter une unité peptidique à partir d'un intermédiaire enzyme-peptide. X représente un alcool, une amine ou un autre type de groupement activant : le réactif est un ester, une amide (peptide) ou un acide carboxylique activé. La vitesse relative de formation du peptide par rapport à celle d'hydrolyse dépend :
|
|
3ème exemple : les enzymes qui catalysent les réactions au voisinage de l'équilibre dans la glycolyse sont les mêmes dans la néoglucogénèse. Attention : en ce qui concerne les réactions très exergonique (ΔG' < 0), la cellule met en place des réactions spécifiques de chaque voie métabolique catalysées par des enzymes spécifiques afin de catalyser la réaction dans un sens ou dans l'autre sens (exemple ci-dessous : les enzymes de la glycolyse versus celles de la néoglucogénèse). |
|||
| enzymes de la glycolyse <=== | ===> enzymes de la néoglucogénèse | ||
hexokinase |
glucose + ATP -> glucose-6-phosphate + ADP | glucose-6-phosphatase |
glucose-6-phosphate + H2O -> glucose + Pi |
phosphofructokinase |
fructose-6-phosphate + ATP -> fructose-1,6-bisphosphate + ADP | fructose-1,6-bisphosphatase |
fructose-1,6-bisphosphate + H2O -> fructose-6-phosphate + Pi |
pyruvate kinase |
phosphoénolpyruvate + ADP -> pyruvate + ATP |
pyruvate carboxylase PEP carboxykinase |
pyruvate + HCO3- + ATP -> oxaloacétate + ADP + Pi oxaloacétate + GTP -> phosphoénolpyruvate + GDP + CO2 |
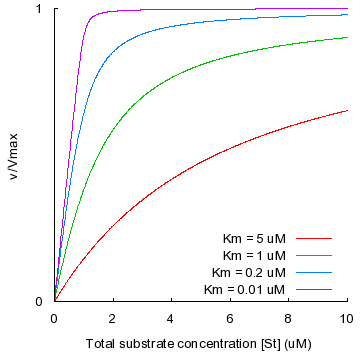
c. La vitesse maximale de la réaction Vmax La vitesse maximale d'une réaction enzymatique est une vitesse initiale théorique : c'est l'asymptote de l'hyperbole obtenue lorsque l'on reporte : vi = f([S0]), que l'on appelle la courbe de saturation. Elle ne peut donc être physiquement mesurée puisqu'elle correspond à une concentration de substrat infinie. Sa valeur ne peut-être qu'approchée :
Remarque 1 : on appelle courbe de saturation la représentation vi = f([S0]) parce que plus la concentration initiale du substrat est élevée, plus il y a de molécules d'enzyme qui fixent une molécule de substrat : l'enzyme est saturée par le substrat. Remarque 2 : quelle que soit la concentration du substrat, [S0] >> [E0]. En conséquence, même si elle n'est pas saturante, la concentration du substrat est toujours en excès. Remarque 3 : Vmax peut être calculée à l'aide de la relation Vmax = kcat . [E0]. La valeur de kcat (et celles des constantes de vitesse de manière générale) peut-être déterminée par des techniques physico-chimiques (par exemple, l'échange isotopique ou l'effet des isotopes). |
d. La constante de dissociation KS et la constante de Michaelis KM
Cette différence traduit deux mécanismes très différents. Dans l'hypothèse du quasi-équilibre, la valeur de la constante de vitesse kcat est très inférieure à k-1. Le complexe ES se dissocie donc plus fréquemment en relarguant le substrat libre qu'il ne subit l'acte catalytique. L'affinité est l'aptitude qu'ont deux molécules à s'associer et KS est une constante de dissociation donc l'inverse d'une constante d'association. En conséquence, KS traduit véritablement l'affinité d'une enzyme pour un substrat. Voir un complément sur le quasi-équilibre Dans l'hypothèse de l'état stationnaire, le complexe ES se dissocie aussi souvent en E et S qu'il est transformé en produit.
KM est donc la concentration en substrat pour laquelle la vitesse initiale mesurée est égale à la moitié de la vitesse initiale maximale Vmax. |
|
e. Le rapport (kcat / KM) ou constante de spécificité 1er cas : une enzyme qui fixerait "facilement" un substrat même à de faibles concentrations (KM faible) pourrait catalyser très lentement la réaction (kcat faible). 2ème cas : inversement, une enzyme qui ne serait saturée qu'à de fortes concentrations de substrat (KM élevé) pourrait accomplir trés souvent l'acte catalytique (kcat élevée). On voit donc que l'un ou l'autre de ces deux paramètres ne permettent pas de caractériser l'efficacité réelle d'une enzyme. Il faut à la fois une fixation du substrat à de faibles concentrations et une catalyse fréquente : c'est donc le rapport (kcat / KM) qui reflète l'efficacité d'une enzyme à catalyser une réaction*. On remarque que la valeur maximale du rapport (kcat / KM) :
La limite du rapport (kcat / KM) est donc la constante de vitesse d'association [enzyme - substrat], k1. La constante k1 a elle-même pour limite la vitesse de diffusion des macromolécules dans le milieu réactionnel (108 - 109 M.s-1). C'est le cas, par exemple, de la triose phosphate isomérase de la glycolyse. |
| Enzyme | KM (M) | kcat (s-1) | kcat / KM (M-1. s-1) |
| Chymotrypsine | 1.5 × 10-2 | 0.14 | 9.3 |
| Pepsine | 3.0 × 10-4 | 0.50 | 1.7 × 103 |
| Tyrosyl-tRNA synthétase | 9.0 × 10-4 | 7.6 | 8.4 × 103 |
| Ribonucléase | 7.9 × 10-3 | 7.9 × 102 | 1.0 × 105 |
| Anhydrase carbonique | 2.6 × 10-2 | 4.0 × 105 | 1.5 × 107 |
| Fumarase | 5.0 × 10-6 | 8.0 × 102 | 1.6 × 108 |
*Pour tenir compte de l'ajustement de la conformation de l'enzyme induit par la fixation du substrat, certains auteurs expriment la constante de spécificité de la façon suivante : K1 . k2, soit le produit de la constante de vitesse du deuxième ordre (fixation du substrat et formation de ES) par la probabilité que, une fois fixé, le substrat le reste et forme le produit. Johnson K.A. (2008) "Role of induced fit in enzyme specificity: a molecular forward/reverse switch" J Biol Chem. 283, 26297-26301 |
|
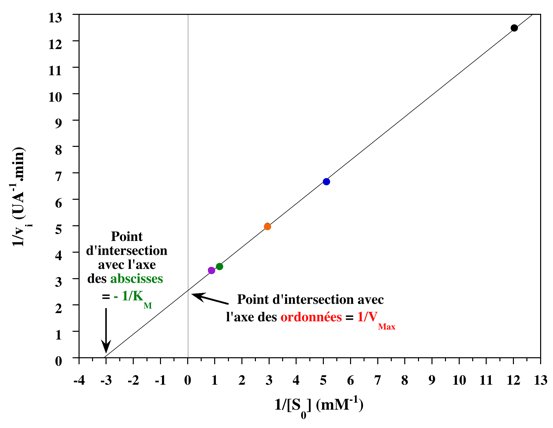
8. Les représentations graphiques - Les méthodes modernes par calculs Les représentations graphiques sont importantes du point de vue didactique. Elles sont faciles à faire avec un crayon, une règle et une feuille de papier. Le principe de ces représentations graphiques est de transformer l'équation de Briggs - Haldane (ou de Henri, Michaelis et Menten), pour 1 ou plusieurs substrats, en présence ou non d'inhibiteurs, afin d'obtenir l'équation d'une droite. a. Représentation de Lineweaver & Burk dite des doubles inverses. Hans Lineweaver & Dean Burk (1934) "The Determination of Enzyme Dissociation Constants" J. Am. Chem. Soc. 56, 658 - 666
On obtient une droite de pente : (KM / Vmax) Dont l'ordonnée à l'origine est : 1 / Vmax et le point d'intersection avec l'axe des abscisses est : - 1/ KM
Voir la procédure pour tracer la droite des doubles inverses. |
b. Représentation de George Eadie & Baren Hofstee : vi = f (vi / [S0])
Cette représentation est aussi appelée représentation de Woolf–Eadie–Augustinsson–Hofstee. c. Représentation de Charles Hanes & Barnet Woolf : [S0] / vi = f ([S0])
Inconvénient : les variables reportées sur les axes ne sont pas indépendantes. d. Représentation de George Eadie & George Scatchard : vi / [S0] = f (vi)
Inconvénient : les variables reportées sur les axes ne sont pas indépendantes. e. Représentation de Eisenthal & Cornish-Bowden : graphe linéaire direct (1974) Robert Eisenthal & Athel Cornish-Bowden (1974)"The direct linear plot. A new graphical procedure for estimating enzyme kinetic parameters" Biochem. J. 13, 715 - 720
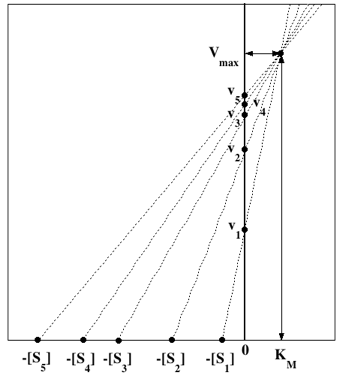
L'axe des abscisses correspond à KM et l'axe des ordonnées correspond à Vmax.
Leur point de concourrance a pour coordonnées KM et Vmax, respectivement.
f. Représentation de Dixon (en présence d'inhibiteurs) : 1 / vi = f ([I0]) pour chaque concentration en substrat. Dixon, M. (1953) "The determination of enzyme inhibitor constants" Biochem J. 55, 170 - 171 Elle est utilisée quand l'interaction entre l'enzyme et l'inhibiteur est plus complexe qu'une inhibition compétitive ou une inhibition non-compétitive classiques. |
g. Les méthodes modernes par calculs et ordinateur L'équation de Henri, Michaelis & Menten ou l'équation de Briggs-Haldane reposent sur des approximations qui, bien que pleinement justifiées, ne sont cependant pas systématiquement respectées in vivo :
Les équations différentielles ne peuvent alors pas être simplifiées. La recherche de leurs solutions est plus complexe et nécessite des méthodes d'analyse numérique, bien souvent par itération (exemples : méthodes de dichotomie, méthode de Newton–Raphson, méthodes de Runge-Kutta, package "nlstools" / R, ...). Des programmes de calcul, basés sur l'intégration numérique des équations différentielles, permettent d'analyser directement les données de cinétiques (enzymatiques ou pas). Exemples : Kaleidagraph, Kinsim, DynaFit, Simfit, KinLSQ, FluSim, ... Plus récemment, la fonction W de Lambert (ou fonction omega) a été utilisée pour analyser les valeurs des paramètres cinétiques :
Articles :
|
h. Exemples de méthodes issues de l'apprentissage profond s'appuyant sur d'immenses jeux de données Voir un cours sur l'apprentissage profond appliqué à la biologie. α. Le modèle UniKP : cadre de prédiction des paramètres cinétiques de réactions enzymatiques (kcat, Km, kcat/Km) basé sur un modèle de langage pré-entraîné ("framework based on pretrained language models") à partir de la séquence d'une enzyme et d'une structure de substrat. Il tient également compte de facteurs environnementaux (notamment pH et température).
β. Service WEB EnzymeML développé avec le langage de balisage XML.
γ. EzMechanism : outil de prédiction des mécanismes d'un site actif pour une réaction enzymatique donnée.
|
Bibliographie
|
![]()