| Application de l'intelligence artificielle et de l'apprentissage profond à quelques domaines de la biologie |
| Tweet |
|
|
1. Préambule 2. Introduction a. Généralités 3. Eléments de vocabulaire et concepts 4. Les différents types d'apprentissage en intelligence artificielle 5. Neurone artificiel : les principes de base a. Le neurone formel - McCulloch & Pitts 6. L'apprentissage profond ("deep-learning") a. Introduction |
d. Les fonctions d'activation 7. Apprentissage profond et structure des protéines a. Apprentissage profond appliqué à la représentation statistique des protéines 8. IA et données de génomique et de transcriptomique 9. IA et quelques exemples en médecine a. Généralités 10. Apprentissage profond, langage naturel et dépense énergétique 11. Liens Internet et références bibliographiques |
1. Préambule L'intelligence artificielle ou IA a de multiples facettes :
Récente (dans ses applications) donc encore en gestation, mal connue des non spécialistes, l'IA fait l'objet de "fantasmes", par exemple dans le domaine de la médecine personnalisée ou de la sécurité des êtres humains (voir les mises en garde de S. Hawking, B. Gates, E. Musk et d'autres personnes). L'IA et l'éthique L'IA et les systèmes d'aide sont confrontés aux biais conscients et inconscients du jugement humain : les données des jeux d'entraînement, les algorithmes et certains choix conceptuels à l'origine des systèmes d'IA peuvent refléter et amplifier les préjugés et les inégalités sociales, culturelles. Il en existe des exemples : reconnaissance vocale qui n'entend pas les femmes, modèles de langage naturel qui font des associations stéréotypées, limite floue entre reconnaissance faciale dans un but de sécurité et atteinte à la vie privée… Lorsque l'apprentissage automatique est intégré dans des systèmes sociaux complexes tels que la justice, les diagnostics de santé, les admissions académiques, l'embauche et la promotion, il peut renforcer les inégalités existantes. Définition "simple" de l'IA L'association des mots "intelligence" et "artificielle" est en soi un élément qui génère un débat et amène chacun à les (re)définir (en particulier, le premier). Une vision relativement "simple" de l'IA pourrait être :
|
2. Introduction L'IA est une science dans laquelle les modèles mathématiques et l'informatique sont omniprésents (quel que soit le domaine d'application). Dans le cas de la biologie, l'IA s'appuie largement sur les ressources et les techniques de la bioinformatique. De plus en plus d'applications de l'IA sont développées, en particulier en robotique, en électronique, en communication et surtout dans le traitement de texte (exemple, Wikipedia ORES) et dans la reconnaissance et l'analyse d'images et de la voix (exemple, Google Duplex). Dans les domaines liés aux sciences biomédicales, on observe une augmentation constante du nombre d'articles de recherche axés sur les applications assistées par l'IA dans des domaines tels que la découverte de médicaments, l'imagerie, le diagnostic, la génétique, la psychologie et les sciences cognitives. Les applications cliniques encourageantes de l'apprentissage automatique pour la prédiction ou le diagnostic de maladies (cancer, maladies cardiovasculaires, rénales ou troubles génétiques) permettent d'envisager un apport très significatif de l'IA. Un nouveau journal de la série des journaux de la collection Nature a vu le jour en janvier 2019 : Nature Machine Intelligence.
Source : Nature Certaines sources mentionnent une augmentation des investissements de plus de 17 milliards de dollars depuis 2009 qui devrait atteindre 36,8 milliards de dollars d'ici 2025. Les principales sociétés biopharmaceutiques ont établi des collaborations avec des sociétés spécialisées dans l'IA. Par exemple :
Enfin, en regard des promesses de profits et s'appuyant sur les jeux de données sans équivalent dont elles disposent, les entreprises majeures du Web se sont emparées de ce domaine et proposent, par exemple, de nombreux services d'analyse d'apprentissage profond ("deep-learning"). |
b. Les processeurs et les puces Figure ci-dessous
Source : Arute et al. (2019)
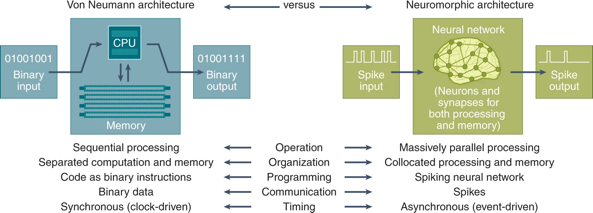
Figures ci-dessous : les puces informatiques neuromorphes sont censées imiter l'architecture de réseau des neurones des cerveaux biologiques.
Source : puce neurosynaptique de IBM
Source : comparaison des architectures von Neumann et neuromorphe (Schuman et al., 2022). Figure ci-dessous
Source : Abu-Hassan et al. (2019) & University of Bath |
c. Environnements pour développer des programmes d'apprentissage profond De tels environnement permettent à toute personne ayant des notions de programmation dans tel ou tel langage - Python étant privilégié - d'écrire des scripts pour l'analyse de ses jeux de données.
Certaines infrastructures logicielles ("frameworks") sont développés par les GAFAM qui obtiennent ainsi gratuitement des jeux de données d'apprentissage colossaux fournies "volontairement" par les internautes. et sont des infrastructures logicielles ("frameworks") pour écrire des programmes dédiés à l'apprentissage machine.
|
3. Eléments de vocabulaire et concepts Réseau de neurones ("neural network") : modèle qui contient au moins une couche dite "cachée". Un réseau de neurones profond contient plusieurs couche cachées. Neurone artificiel ("artificial neuron") : fonction mathématique simple qui prend en entrée un vecteur de valeurs réelles et calcule la moyenne pondérée de ces valeurs suivie d'une transformation non linéaire. Poids ("weight") : ce sont les paramètres du neurone qui pondèrent les valeurs.
Apprentissage profond ("deep learning") : réseaux de neurones avec plusieurs couches de neurones artificiels. La sortie d'une couche (la sortie d'un neurone) est introduite en entrée dans la couche suivante (un autre neurone) pour obtenir une plus grande flexibilité.
Validation croisée : stratégie d'apprentissage automatique où l'ensemble des données est fractionné plusieurs fois en jeux de données d'entraînement ("training dataset") et de validation. La performance de validation moyenne des multiples groupes est utilisée pour sélectionner le modèle final. Rétro-propagation ("back propagation") : algorithme pour former un réseau de neurones en mettant à jour ses paramètres (ses poids) en utilisant la dérivée de la performance du réseau par rapport aux paramètres. Fonction de regroupement ("pooling") : elle substitue la valeur de sortie d'un réseau à un endroit donné par une valeur statistique des sorties voisines (le plus souvent la valeur maximale, moyenne et médiane). Auto-encodeur : classe de réseaux de neurones qui effectue une réduction de dimensionnalité non linéaire. Sur-ajustement ("overfitting") : le modèle correspond à l'ensemble d'apprentissage mais n'est pas généralisable aux données non montrées. C'est souvent le cas des modèles flexibles avec de nombreux paramètres libres contrairement à ceux qui ont moins de paramètres que le jeu de données d'apprentissage. Filtres : ce sont les paramètres d'une couche convolutive ("convolutional layer"). Dans la première couche d'un réseau convolutif de séquences (de nucléotides par exemple), elles peuvent être interprétées comme des matrices de pondération de position de ces nucléotides. Fonction d'activation : fonction mathématique qui calcule la valeur de sortie d'un neurone artificiel. Fonction sigmoïde : exemple d'une fonction d'activation de certains réseaux de neurones. Elle permet la conversion de valeurs brutes issues d'une régression logistique ou multinomiale en probabilité. Transformateur ("transformer") : modèle d'apprentissage profond qui utilise le processus d'auto-attention. Ce type de modèle pondère différemment l'importance de différentes parties des données en entrée : il améliore ainsi certaines parties et en diminue d'autres. Il est trés utilisé dans le traitement du langage naturel (exemple : ProteinBERT). |
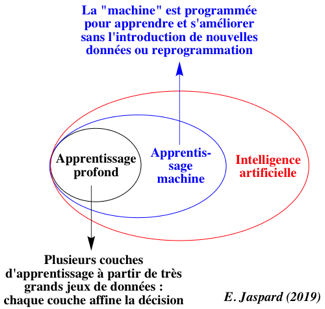
4. Les différents types d'apprentissage en intelligence artificielle Intelligence artificielle : discipline scientifique qui tire ses racines principalement de la philosophie, de la psychologie, des mathématiques et de l'informatique et qui vise à comprendre et à développer des systèmes affichant les "propriétés de l'intelligence".
Apprentissage machine ("machine learning") : sous-discipline de l'IA où des algorithmes apprennent des associations à partir de jeux de données qui leur servent de jeux de données d'apprentissage.
Source : Walsh et al. (2021) Apprentissage profond ("machine learning") : il repose sur plusieurs couches de représentation de très grands jeux de données avec des transformations successives.
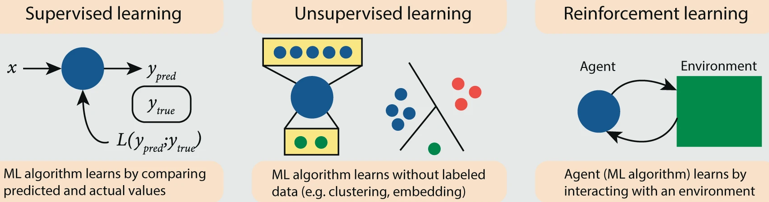
Apprentissage automatique : apprentissage supervisé, apprentissage non supervisé et apprentissage par renforcement.
Source : Sapoval et al. (2022) Quelques théoriciens de l'apprentissage automatique :
Apprentissage par renforcement ("reinforcement learning") : le système apprenant est dans un environnement donné et il apprend à prendre des décisions, de manière autonome, à partir des expériences antérieures afin d'optimiser une récompense quantitative qui évolue. Apprentissage supervisé ("supervised learning") : programme d'apprentissage des associations entre les données en entrée et en sortie par l'analyse des sorties qui présentent un intérêt défini par un superviseur (généralement humain). C'est l'un des domaines prépondérant de l'apprentissage automatique avec de nombreuses applications dans le domaine de la santé. Exemple d'apprentissage supervisé : créer un système qui reconnaisse et classe des images contenant une maison, une voiture ou un animal.
Ces paramètres ajustables (appelés poids) sont des nombres réels : il peut y avoir des centaines de millions de catégories et de poids ajustables à partir desquels la machine (l'algorithme / le programme) est entrainée. Pour ajuster correctement le vecteur de poids, l'algorithme d'apprentissage calcule un vecteur de gradient qui, pour chaque poids, indique de quelle valeur augmente ou diminue l'erreur si le poids est légèrement modifié. Le vecteur de poids est ensuite ajusté dans la direction opposée au vecteur de gradient. Apprentissage non supervisé ("unsupervised learning") : programme qui apprend les associations à partir de jeux de données sans qu'on lui fournisse de définition de ces associations. L'apprentissage non supervisé est capable d'identifier des prédicteurs inconnus. |
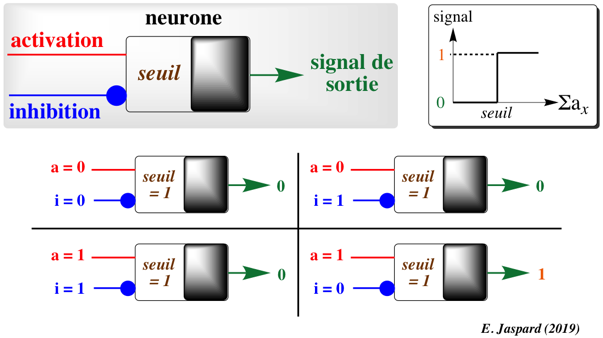
5. Neurone artificiel : les principes de base a. Le neurone formel - McCulloch & Pitts L'article fondateur de la théorie de neurone formel a été publié en 1943 par le neurophysiologiste Warren McCulloch (neurophysiologiste) et le logicien Walter Pitts. L'activité de ce neurone formel suit la loi de "tout ou rien" (Edgar Adrian, Prix Nobel de physiologie ou médecine en 1932) : la force avec laquelle un nerf répond à un stimulus est indépendante de la force de ce stimulus. Ce comportement est parfaitement compatible avec une logique binaire puisque :
Le signal de sortie est une fonction de (ax, iy) :
|
b. Règle de Hebb et poids synaptique entre les neurones Le neuropsychologue Donald Hebb a mis en évidence l'effet du couplage entre synapses dans le processus d'apprentissage. Règle de Hebb ("A neurophysiological postulate", p 62, 1949) : "Lorsqu'un axone de la cellule A est suffisamment proche pour exciter une cellule B et qu'elle participe de manière répétée ou persistante au déclenchement de B, un processus de croissance ou un changement métabolique a lieu dans l'une ou dans les deux cellules, de sorte que l'efficacité de A à déclencher B est augmentée." En ce qui concerne les neurones artificiels, la règle de Hebb permet de déterminer comment modifier les poids entre 2 neurones (neurone j -> neurone i) : Δwi,j = xi.yj
La règle d'apprentissage la plus simple est : wi,j(t+1) = wi,j(t) + η.xi(t).yj(t)
Si on initialise les poids à 0 et le pas d'apprentissage à 1, après k exemples d'apprentissage on obtient : wi,j = ∑k xi,k.yj,k |
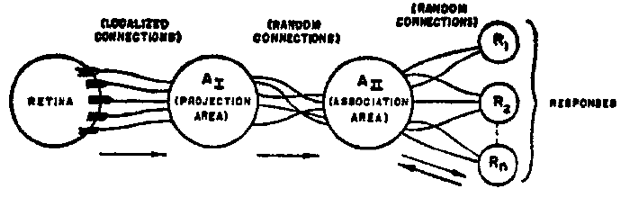
c. Le Perceptron - Frank Rosenblatt C'est le premier modèle de réseau capable d'apprendre. Il a été créé en 1958 par le psychologue Frank Rosenblatt. L'organisation d'un photoperceptron (Perceptron répondant à des motifs optiques sous forme de stimuli) est la suivante :
Source : Rosenblatt, F. (1958)
Les "A-units" et les "R-units" sont des neurones formels de type McCulloch – Pitts mais leurs synapses sont variables et s'adaptent en conséquence selon la règle de Hebb. Cependant la règle de Hebb :
Le Perceptron est constitué :
La règle du Perceptron est : wi,j(t+1) = wi,j(t) + η.(xs - xi(t)).yj Voir : "19-line Line-by-line Python Perceptron". |
d. Théories et modèles plus récents De nombreuses théories ont ensuite été développées (essentiellement à partir des années 1980). Elles ont aboutit à des règles ou des modèles qui décrivent les propriétés des différents types d'apprentissage (supervisé, non supervisé, ...). Par exemple :
Source : "Bioengineering Community" (Nature) |
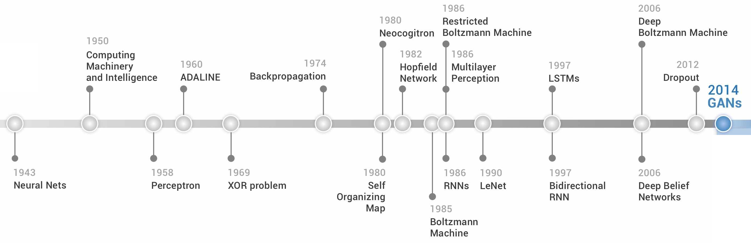
6. L'apprentissage profond ("deep-learning") L'apprentissage profond permet aux modèles informatiques composés de plusieurs couches de traitement d'apprendre des représentations des données avec plusieurs niveaux d'abstraction. L'apprentissage profond permet de découvrir des "structures complexes" au sein de grands ensembles de données en utilisant la rétro-propagation (par exemple) qui dicte à la machine comment celle-ci doit modifier ses paramètres internes pour calculer la représentation de chaque couche à partir de la représentation de la couche précédente. Figure ci-dessous : chronologie du développement des algorithmes d'apprentissage profond et d'apprentissage automatique.
Source : Cao et al. (2018) NN : "Neural Network", réseau de neurones - BP : "back propagation" - DBN : "deep belief network" - SVM : "support vector machine" (machine à vecteurs de support) - AE :"auto-encoder" - VAE :"variational auto-encoder" - GAN : "generative adversarial network" - WGAN : "Wasserstein GAN". |
b. Principe de fonctionnement et architectures des réseaux de neurones Un réseau de neurones permet de résoudre de nombreux problèmes d'intelligence artificielle. Schématiquement, le fonctionnement est le suivant :
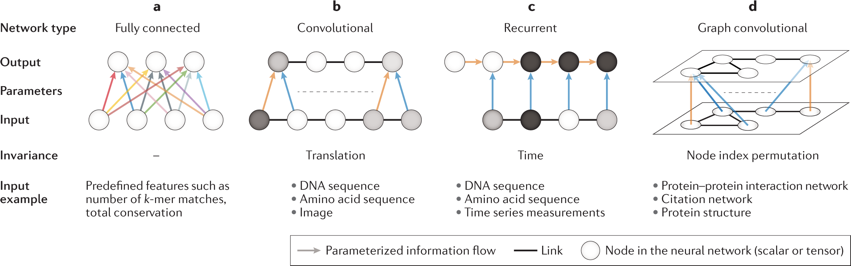
Les architectures des réseaux de neurones peuvent être classés selon leurs systèmes de connectivité et de partage des paramètres :
Source : Eraslan et al. (2019) Figure ci-dessus :
a. Les couches entièrement connectées ("fully connected layers") supposent que les entités en entrée n'ont pas d'ordre particulier. Elles appliquent donc des paramètres différents pour différentes entités en entrée. b. Les couches convolutives ("convolutional layers") ou réseaux convolutifs supposent que des sous-ensembles locaux d'entités en entrée (par exemple, les nucléotides consécutifs dans une séquence d'ADN) représentent des motifs. Un réseaux de neurones convolutif ("Convolutional Neural Network" - CNN) utilise la convolution à la place d'une multiplication de matrices dans les couches cachées ("hidden layers"). Un CNN typique est constitué de couches denses entièrement connectées et de couches convolutives. c. Les couches récurrentes ("recurrent layers") supposent que les entités en entrée doivent être traitées séquentiellement et que l'élément de séquence dépend de tous les éléments de séquence précédents. La même opération est appliquée à chaque élément de séquence (flèches bleue et orange dans la figure ci-dessus) et les informations de l'élément de séquence suivant sont incorporées dans la mémoire (flèches orange) et reportées. d. Les graphes convolutifs supposent que la structure des entités en entrée suit celle d'un graphe connu. Le même ensemble de paramètres est utilisé pour traiter tous les nœuds et impose ainsi une invariance à la commande des nœuds. |
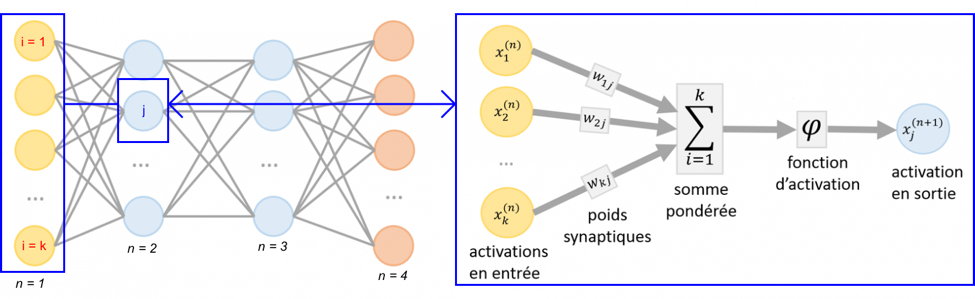
c. Exemple d'apprentissage supervisé d'un réseau de neurones qui classe des images des chiffres 0 à 9
Source : Yamashita et al. (2018) Couche d'entrée (n = 1) de ce réseau simple (figure ci-dessous) :
Les couches de sortie (n = 2, 3, ...) sont constituées de neurones yj avec j = 0 à 9, les 10 chiffres correspondant aux images du jeux de données d'entraînement.
Source : "Deep-learning pas à pas" (SQLI) Les niveaux d'activation des neurones sont calculés de manière récursive, couche par couche : ceux de la couche de neurones n+1 sont calculés avec une fonction d'activation φ sur la base des niveaux d'activation des neurones de la couche n pondérés par un poids wi,j. La somme est effectuée sur tous les neurones i de la couche n connectés au neurone j de la couche n+1 : xj=0,9(n+1) = φ [ ∑i=1,k (wi,j . xi(n)) ]
|
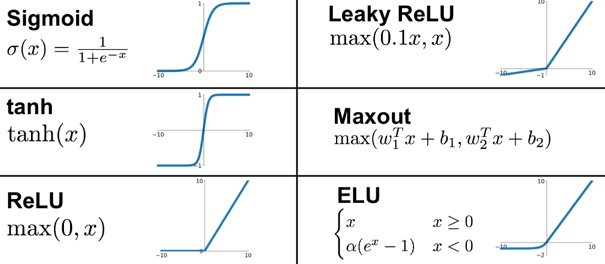
Exemples de fonctions d'activation (représentées par g(x) avec x = valeur de la couche d'entrée) : Sigmoïde
Tangente hyperbolique
Softmax
Unité linéaire rectifiée ("Rectified Linear Unit", ReLU)
Variantes de ReLU
Maxout
Source : Towards Data Science Voir un large éventail de fonctions d'activation et leurs caractéristiques. |
e. Optimisation et fonction objectif (fonction de perte) Les réseaux de neurones sont formés par un processus d'optimisation nécessitant une fonction de perte qui calcule l'erreur du modèle : cette fonction mesurent la différence entre la sortie du réseau (obtenue avec les paramètres du modèle f(x|θ)) et le résultat attendu. Exemple de résultat attendu : les étiquettes de classe "VRAI" dans les tâches de classification ou le niveau réel dans les tâches de prédiction.
import numpy as np def Erreur_Quadratique_Moyenne(y_predit, y): Carre_Erreur = (y_predit - y)**2 Somme_Carre_Erreur = np.sum(Carre_Erreur) ErrQuadMoyen = Somme_Carre_Erreur / nombreY return(ErrQuadMoyen) Perte d'entropie croisée ("Cross Entropy Loss") La perte d'entropie croisée mesure les performances d'un modèle de classification. La sortie est une valeur de probabilité comprise entre 0 et 1. La perte d'entropie croisée augmente à mesure que la probabilité prédite diverge de l'étiquette réelle.
Exemple de code Python pour le calcul de la perte d'entropie croisée : import numpy as np predictions = np.array([[0.25,0.25,0.25,0.25],[0.01,0.01,0.01,0.96]]) targets = np.array([[0,0,0,1],[0,0,0,1]]) def cross_entropy(predictions, targets, epsilon=1e-10) : predictions = np.clip(predictions, epsilon, 1. - epsilon) N = predictions.shape[0] ce_loss = -np.sum(np.sum(targets * np.log(predictions + 1e-5)))/N return ce_loss cross_entropy_loss = cross_entropy(predictions, targets)
print ("Cross entropy loss is: " + str(cross_entropy_loss))
|
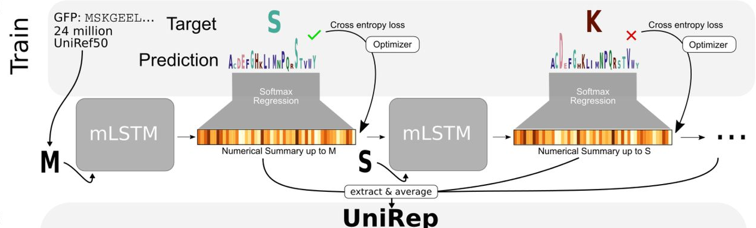
7. Apprentissage profond et structure des protéines a. Apprentissage profond appliqué à la représentation statistiques des protéines Le modèle de représentation unifiée UniRep ("Unified Representation") a été élaboré :
Le modèle a été construit sur la base d'une architecture mLSTM - RNN (multiplicative Long-Short-Term-Memory Recurrent Neural Network) :
Une fois entraîné, le modèle génère (sans données liées à la structure ou à l'évolution) un seul vecteur de longueur fixe représentant la séquence d'acides aminés en entrée.
Source : Alley et al. (2019) |
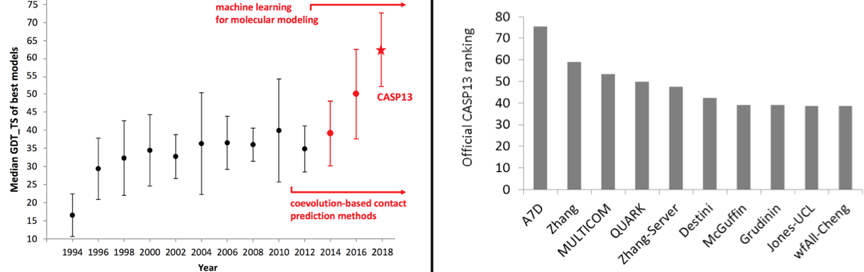
b. Apprentissage profond appliqué à la prédiction de la structure 3D des protéines Depuis 1994, des biologistes spécialisés en calcul informatique participent à une expérience communautaire mondiale visant à évaluer les techniques de prédiction de la structure des protéines. Ce concours, appelé CASP ("Critical Assessment of protein Structure Prediction"), a lieu tous les 2 ans et aboutit à une conférence plénière.
Source : Swiss Institute of Bioinformatics
CASP permet donc de faire le point sur l'état actuel de la prédiction de la structure des protéines, en identifiant les progrès accomplis au cours des deux années écoulées et en soulignant les domaines dans lesquels les efforts futurs pourraient être les plus productifs. Les développements récents incluent, par exemple, les réseaux adverses génératifs ("Generative Adversarial Network", GAN, réseaux non-supervisés), qui apprennent à imiter toute distribution de données pour créer des représentations réalistes de l'objet d'intérêt, telles que les conformations locales d'une structure protéique. |
c. Cartes de distances et de contacts pour la prédiction de la structure 3D L'analyse de co-évolution ("residue-residue coevolutionary") est basée sur l'observation selon laquelle 2 résidus d'acides aminés en contact (ou proches dans l'espace selon un seuil de distance, par exemple 8 Å) doivent co-évoluer pour maintenir ce contact. En d'autres termes, si un acide aminé est muté en résidu chargé positivement, l'acide aminé avec lequel il est en contact doit être muté en résidu chargé négativement. Depuis le concours CASP11 et CASP12, les scores de co-évolution sont de plus en plus utilisés comme entrées des méthodes d'apprentissage automatique afin d'améliorer la prédiction des contacts et des distances ("inter-residue contacts and distances"). |
| Exemples d'outils de prédiction de la structure 3D des protéines | |
| Outils | Principe |
| CASP13 | Les prédicteurs ont calculé la topologie de repliement de la chaîne polypeptidique avec une résolution < 3 Å par rapport à la structure déterminée expérimentalement. |
| AlphaFold | Conceptuellement, AlphaFold était à l'origine un réseau de neurones pour prédire les distances probables entre les paires d'acides aminés et les angles de chaque liaison peptidique reliant les résidus d'acides aminés. Ces 2 prédictions ont ensuite été intégrées à un score avec le "score2" du logiciel de modélisation Rosetta.
|
| RaptorX - Contact | Serveur Web qui applique un réseau de neurones convolutifs ultra-profond pour la prédiction des cartes de contacts et la prédiction de matrices de distances de séquences protéiques sans l'utilisation de modèles. RaptorX utilise uniquement la moyenne et la variance de la distribution prédite. |
| iFeature | Package Python et un serveur Web pour l'extraction et la sélection de caractéristiques à partir de séquences de protéines et de peptides. |
| iLearn | Plate-forme intégrée et "méta-apprenant" pour l'ingénierie des caractéristiques, l'analyse par apprentissage automatique et la modélisation des données de séquences d'ADN, d'ARN et de protéines. |
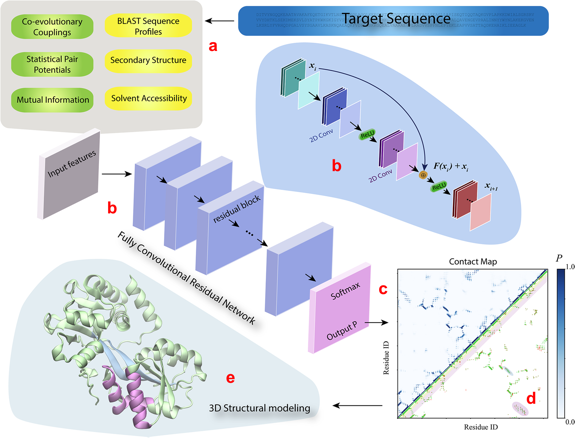
| DESTINI | (a) Les caractéristiques 1D (bulles jaunes en haut à gauche, figure ci-dessous) et 2D (bulles verts) de la séquence de la protéine analysée ("Target sequence") sont extraites à partir des données génomiques et structurales. (b) Ces caractéristiques constituent les entrées ("Input features") d'un réseau convolutif composé de plusieurs blocs résiduels identiques (le détail de l'architecture est décrit dans la bulle bleue claire). (c) La couche de sortie du réseau est une couche d'activation Softmax qui génère le score de probabilité ("Output P") pour chaque paire de résidus de la séquence cible. (d) La sortie est affichée sous forme d'une carte de contacts prédits ("Contact map") avec les scores de probabilité (partie gauche - haute de la carte) et une comparaison des contacts prédits (P > 0,5) avec les contacts véritables (partie droite - basse). (e) Les contacts prédits sont ensuite utilisés pour générer le modèle 3D de la protéine analysée. Les couleur des contacts entre les hélices α et les brins β sont identiques à celles de la carte des contacts (d).
Source : Gao et al. (2019) |
| MULTICOM | Il s'appuie sur 3 concepts principaux : (i) la prédiction de la distance de contacts avec des réseaux de neurones convolutifs; (ii) la modélisation sans modèle (ab initio) dirigée par la distance; (iii) le classement du modèle de la protéine renforcé par l'apprentissage profond et la prédiction des contacts. |
| SPIDER2 | Prédiction des erreurs dans les valeurs prédites des angles de torsion de la chaîne polypeptidique, de l'accessibilité au solvant, des contact . |
| Early Folding Residue Predictor | Le repliement des protéines est un processus qui s'effectue par étapes : certains résidus d'acides aminés semblent initier ce processus ("early folding residues"). Certaines données expérimentales concernant ces résidus sont fournies par la base de données Start2Fold mais pour la plupart des structures protéiques, aucune donnée n'est disponible. Early Folding Residue Predictor prédit ("Generalized Matrix Learning Vector Quantization") la position de ces résidus à partir de structures protéiques (PDB). |
Autres outils : UniCon3D (CASP11) - DNCON2 (CASP 12) - MetaPSICOV2 (CASP 12, utilise les données de PDB70 et Uniref100) - DeepCov, ... |
|
d. Apprentissage profond, langage naturel et prédiction de la structure des macromolécules biologiques Un modèle de langage naturel large ou statistique ("large language model") est basé sur un réseau de neurones d'apprentissage profond (exemple de l'architecture "Transformer" - voir ci-dessous) et formé à partir d'un corpus de données textuelles gigantesque (des centaines de milliards de mots et/ou de phrases).
|
| Programme | Création | Entreprise | Paramètres (milliards) |
Divers |
| WuDao 2.0 | 2021 | Beijing Academy of Artificial Intelligence | 1750 | ----- |
| MT-NLG | 2022 | Nvidia | 530 | "Megatron-Turing Natural Language Generation model" |
| Bloom | 2022 | BigScience | 176 | "BigScience Large Open-science Open-access Multilingual Language Model" |
| GPT-3 | 2020 | OpenAI | 175 | Transformer "Generative Pre-Trained" |
| LaMDA | 2021 | 137 | ----- | |
| ESMFold ("Evolutionary Scale Modeling") | 2022 | Meta AI | 15 | "ESM Metagenomic Atlas" : plus de 600 millions de structures tridimensionnelles de protéines. Voir le code source (GitHub) |
| BERT | 2018 | 0,34 |
Les méthodes basées sur l'apprentissage profond et le langage naturel sont désormais appliquées à la détermination de la structure des ARN : voir Pearce et al., 2022. EpiCas-DL : programme de prédiction par apprentissage profond de l'activité de l'ARN guide simple ("single guide RNA") pour l'édition de l'épigénome médiée par CRISPR. scMDC ("Single Cell Multimodal Deep Clustering") : méthode d'apprentissage profond pour le regroupement de données multi-omiques unicellulaires ("single-cell multi-omics data clustering"). L'architecture réseau de scMDC utilise un auto-encodeur multimodal. |
8. IA et données de génomique et de transcriptomique Voir un ensemble de cours sur la génomique et la protéomique. |
| Exemples d'outils pour l'analyse de données issues de certains domaines en omique | |
| Outils | But |
| Deepnet-rbp | Cadre d'apprentissage profond pour modéliser les préférences de fixation des protéines de liaison à l'ARN ("RNA-binding proteins") en intégrant la séquence primaire, les profils de structures secondaires et de structures tertiaires prédits des sites cibles. |
| DeepBind | Prédiction des sites de fixation des protéines de liaison à l'ADN et à l'ARN. |
| DeepRibo | Réseau de neurones pour l'annotation de gènes de procaryotes basé sur une combinaison de profils signature des ribosomes et de profils de sites de fixation. |
| PEDLA | Apprentissage profond pour l'identification des régions cis-régulatrices. |
| LeNup | Réseau de neurones convolutif pour l'apprentissage du positionnement des nucléosomes à partir de séquences d'ADN. |
| DNN-HMM | Identification de novo des régions d'initiation et de terminaison de l'ADN qui se répliquent plus tôt ou plus tard ("early or late replication domain") que leurs voisines en amont et en aval ("up or down transition zone"). Réseaux de neurones et Modèle de Markov caché. |
| Basset | Réseau de neurones convolutif qui, sur la base d'un seul séquençage d'un type de cellule, permet :
|
| DeepCpG | Prédiction des méthylations de l'ADN (épigénomique). |
| deepTarget | Réseau de neurones récurrent pour la prédiction de cible de miRNA (interférence ARN). |
| DeepMirTar | Réseau de neurones récurrent pour la prédiction de cible de miRNA de l'homme. |
| Interactomique | Prédiction des interactions protéine-protéine par intégration d'un modèle de classement de type forêt d'arbres décisionnels par rotation des caractéristiques sélectives ("Feature-selective rotation forest classifier" - FSRF) à un réseau de neurones convolutifs.
La démarche contient 3 étapes :
|
| DN-Fold | Système de reconnaissance du repliement des protéines. |
| MusiteDeep | Prédiction des sites de phosphorylation généraux et spécifiques des kinases. |
| DeepLoc | Prédiction de la localisation subcellulaire des protéines. |
| biovec 0.2.7 | Méthodes d'extraction des caractéristiques des séquences biologiques et méthodes de représentation des séquences biologiques utilisées dans les applications d'apprentissage profond en génomique et en protéomique :
|
| EnzyNet | Prédiction de la classification E.C ("Enzyme Commission") des enzymes avec des réseaux de neurones convolutifs. |
| DeepCSeqSite | Prédiction des résidus d'acides aminés impliqués dans la liaison protéine-ligand avec des réseaux neuronaux convolutifs. |
| AMPSphere (AntiMicrobial Peptides) |
L'apprentissage automatique prédit plusieurs centaines de milliers de nouveaux antibiotiques codé par l'ensemble du microbiome mondial séquencé. |
9. IA et quelques exemples en médecine Les progrès des technologies biologiques et médicales nous fournissent des volumes colossaux de données biologiques et physiologiques (images médicales, électro-encéphalographie, séquences de génomes, de protéines, ... ). Apprendre de ces données augmente la compréhension des maladies donc de la santé humaine. Développés à partir de réseaux de neurones artificiels, les algorithmes basés sur l'apprentissage profond et l'optimisation des modèles sont prometteurs pour l'extraction de caractéristiques et de modèles d'apprentissage à partir de données complexes. Ces méthodes peuvent être appliquées à la classification d'images médicales, à l'analyse de génomes, à la prédiction de la structure des protéines, ... L'IA peut être utilisée de différentes manières en médecine. En voici 4 exemples : Outil d'annotation des données cliniques : environ 80% des données de soins sont non structurées. Or les méthodes d'IA peuvent lire des données non structurées via leur aptitude à "comprendre" (traiter) le langage naturel. Ces méthodes peuvent donc lire des textes cliniques de n'importe quelle source puis identifier, catégoriser et coder des concepts médicaux et sociaux. Informations sur les données des patients : l'IA peut identifier des problèmes dans l'historique des dossiers médicaux des patients (texte structuré et non structuré). Certaines méthodes résument l'historique des soins liés à ces problèmes en un document cognitif des données d'un patient. Similitude de patient : l'IA peut mesurer la "similarité clinique" entre patients. Cela permet aux chercheurs de créer des cohortes dynamiques de patients plutôt que des cohortes statiques et cela permet de déterminer le protocole de soins le mieux adapté à un groupe de patients donné. Hypothèses médicales : les technologies d'IA permettent aux chercheurs de trouver des informations dans des publications médicales non structurées et d'identifier les documents liés sémantiquement à toute combinaison de concepts médicaux. Le registre AIME : plateforme communautaire pour l'IA en biomédecine qui a pour but d'améliorer l'accessibilité, la reproductibilité et la convivialité des modèles d'IA biomédicale. Il permet des révisions futures par la communauté. |
| Domaines médicaux d'application de l'apprentissage profond | ||
| Domaine | Données en entrée | Exemples de thèmes de recherches |
| Domaines en "omique" |
|
|
| Imagerie médicale |
|
|
| Traitement du signal biomédical |
|
|
| Source : Min et al. (2017) | ||
|
La recherche classique de médicaments commence par le test de milliers de petites molécules pour n'en retenir que quelques unes, parmi lesquelles une seule réussit, éventuellement, les essais cliniques sur des patients humains. La découverte de médicaments nécessite donc :
L'IA promet de réduire considérablement les délais et les coûts en facilitant l'identification rapide des composés. Les modèles génératifs profonds sont des techniques d'apprentissage automatique qui utilisent des réseaux de neurones qui peuvent générer des objets dotés de certaines propriétés, telles que l'activité vis-à-vis d'une cible donnée : ces modèles sont donc adaptés à la découverte de médicaments. Un système d'IA appelé apprentissage par renforcement tensoriel génératif ("Generative Tensorial Reinforcement Learning", GENTRL) a permis de concevoir 6 nouveaux inhibiteurs de l'enzyme DDR1 en 21 jours (Zhavoronkov et al., 2019):
La validation in vitro puis in vivo n'aura nécessité que 46 jours. L'enzyme DDR1 ("Epithelial discoidin domain-containing receptor 1") est une tyrosine kinase, récepteur à la surface cellulaire du collagène fibrillaire. Elle régule la fixation des cellules à la matrice extracellulaire, le remodelage de cette matrice, la migration cellulaire, la différenciation, la survie et la prolifération cellulaire. |
| Exemples d'outils informatiques basés sur l'IA pour la découverte de médicaments | |
| Outils | But |
| Chemputer | Format normalisé pour rapporter une procédure de synthèse chimique |
| DeepChem | Outil Python pour des tâches de découverte de médicaments |
| DeepNeuralNet-QSAR | Prédiction d'activité moléculaire |
| DeepTox | Prédiction de toxicité |
| DeltaVina | Une fonction de scores pour ré-évaluer l'affinité de la liaison protéine-ligand |
| Hit Dexter | Modèles pour la prédiction de molécules susceptibles de répondre à des essais biochimiques |
| Neural Graph Fingerprints | Prédiction des propriétés de nouvelles molécules |
| ODDT | Boîte à outils pour la chimio-informatique et la modélisation moléculaire |
| ORGANIC | Outil pour créer des molécules avec des propriétés souhaitées |
| PotentialNet | Prédiction de l'affinité de la liaison de ligands basée sur des graphes de réseau de neurones convolutifs |
| PPB2 | Prédiction polypharmacologique |
| QML | Boîte à outils Python pour l'apprentissage profond quantique |
| REINVENT | Conception moléculaire de novo utilisant un réseau de neurones récurrent ("recurrent neural network") et l'apprentissage par renforcement ("reinforcement learning") |
| SCScore | Fonction de scores pour évaluer la complexité de la synthèse d'une molécule |
| Source : Chan et al. (2019) | |
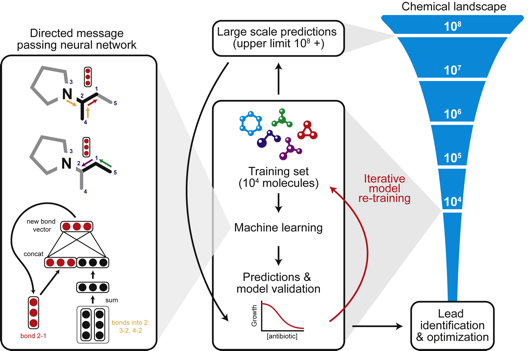
c. Illustration : recherche de nouveaux antibiotiques Un réseau de neurones a été créé pour apprendre à reconnaître les particularités structurales des molécules qui inhibent la croissance d'Escherichia coli BW25113 en utilisant une collection de 2335 molécules à activité anti-croissance bactérienne connue, dont environ 300 antibiotiques et 800 produits naturels d'origine végétale, animale et microbienne (Stokes et al., 2020).
Source : Stokes et al. (2020) Ce modèle a ensuite été utilisé pour analyser plus de 100 millions de molécules de la bibliothèque "Drug Repurposing Hub" (entre autres molécules, cette bibliothèque contient environ 6000 molécules à l'étude pour des maladies chez l'homme) : 120 molécules à activité anti-croissance bactérienne ont été sélectionnées pour des tests. Une molécule (étudiée pour un éventuel traitement du diabète) s'est avérée un antibiotique puissant : l'halicine ("c-Jun N-terminal kinase inhibitor", SU3327), dénommée ainsi d'après HAL (l'ordinateur du film "2001: A Space Odyssey").
Puis plus de 107 millions de structures de molécules de la base de données ZINC15 (base de données gratuite de près d'un milliard de composés disponibles dans le commerce pour le criblage virtuel) ont été analysées : 23 molécules candidates ont été découvertes et testées et 8 molécules ont une une activité antibactérienne. En particulier, deux d'entre elles ont une activité contre un large éventail d'agents pathogènes et contre des souches d'Escherichia coli résistantes aux antibiotiques. |
10. Apprentissage profond, langage naturel et dépense énergétique Les avancées matérielles et théoriques de la [formation / entraînement] de réseaux de neurones permettent d'entraîner des modèles d'apprentissage profond avec des volumes de données gigantesques (par exemple, le corpus Wikipedia). Cependant, l'entraînement et la mise au point de tels réseaux nécessite des ressources très importantes :
Quatre modèles parmi les plus performants dans le domaine du traitement du langage naturel ont été comparés : Transformer, ELMo, BERT et GPT-2.
Chacun de ces modèles a été entraîné avec une seule unité de traitement graphique ("Graphics Processing Unit" - GPU, NVIDIA) pendant une journée au maximum puis la consommation électrique a été mesurée. Le nombre d'heures d'entraînement du modèle indiqué dans les documents originaux a servi de base pour calculer l'énergie totale consommée tout au long du processus d'entraînement. Ce nombre a été converti en livres ("pounds", mesure anglo-saxonne) d'équivalent CO2 sur la base du bouquet énergétique moyen aux États-Unis. |
| Processus | Consommation en livres ("pounds") d'équivalent CO2 |
| Vol d'1 passager AR New-York - San Francisco | 1984 |
| "1 année moyenne" de la vie d'un être humain | 11.023 |
| "1 année moyenne" de la vie d'un américain | 36.156 |
| Côut énergétique de la vie moyenne d'une voiture (carburant inclu) | 126.000 |
| Traitement par BERT | 1438 |
| Traitement par Transformer | 192 |
| Traitement par Transformer avec processus de mise au point (recherche d'architecture neuronale) | 626.155 |
| Source : Strubell et al. (2019) | |
Les résultats montrent que :
En particulier, le processus de mise au point appelé recherche d'architecture neuronale ("neural architecture search"), qui optimise un modèle en modifiant progressivement la conception d'un réseau de neurones par essais et erreurs exhaustifs, a un coût très élevé pour une performance faible : par exemple, sans ce processus l'entraînement de Transformer prend 84 heures et avec ce processus, il prend plus de 270.000 heures (soit 3000 fois plus d'énergie et des mois d'entraînement). |
| 11. Liens Internet et références bibliographiques |
|
"Réseaux Neuronaux" J.-P. Rennard (2006) Vuibert, Paris - ISBN : 2-7117-4830-8 "Cancers, vers une révolution thérapeutique ?" Barbet et al. (2019) ISTE Editions Ltd - ISBN : 978-1-78405-622-3 |
|
The European Lab for Learning and Intelligent Systems Python : Machine Learning Keras : The Python Deep Learning library Towards Data Science |
|
Quantum : Google AI OpenAI Glossaire du "Machine learning" "The AIMe registry for artificial intelligence in biomedical research" |
|
Les technologies digitales décryptées par SQLI The Library of Integrated Network-Based Cellular Signatures Program |
|
AlphaFold DB : système de prédiction de la structure des protéines du protéome humain et de environ 50 autres organismes clés, basé sur l'intelligence artificielle. GDB-13 : base de données d'environ 1 milliard de structures de molécules organiques contenant jusqu'à 13 atomes de C, N, O, S et Cl. AMPSphere (AntiMicrobial Peptides) |
|
Algorithms in Nature Didacticiel interactif pour la création d'un réseau de neurones de convolution permettant de découvrir des motifs de liaison à l'ADN Bengio Y., Hinton G.E. & LeCun Y. : "ACM A.M. Turing Award" 2018 |
|
McCulloch & Pitts (1943) "A Logical calculus of ideas immanent in nervous activity" Bull. Math. Biophys. 5, 115 - 133 Hebb D.O. (1949) "The organization of behavior : a neuropsychological theory" New York : Wiley & Sons Rosenblatt F. (1958) "The perceptron : a probabilistic model for information storage and organization in the brain" Psychol. Rev. 65, 386 - 408 Widrow B. (1960) "An adaptative "Adaline" neuron using chemical "memistors"" Stanford Electronics Lab. Tech. Report 1553-2 Widrow & Hoff, Jr. (1960) "Adaptive switching circuits'' IRE WESCON Convention Record 4, 96 - 104 |
|
Hochreiter & Schmidhuber (1997) "Long Short-Term Memory" Neural Comput. 9, 1735 - 1780 Hinton G.E. (2011) "Machine learning for neuroscience" Neural Syst. Circuits 1, 12 LeCun Y., Bengio Y. & Hinton G.E. (2015) "Deep learning" Nature 521, 436 - 444 |
|
Chakraborty et al. (2017) "Artificial Intelligence in Biological Data" J. Inform. Tech. Softw. Eng. 7, 4 Min et al. (2017) "Deep learning in bioinformatics" Brief. Bioinform. 18, 851 - 869 |
|
Panch et al. (2018) "Artificial intelligence, machine learning and health systems" J. Glob. Health 8, 020303 Chouldechova et al. (2018) "A case study of algorithm-assisted decision making in child maltreatment hotline screening decisions" Proceed. 1st Conf. Fairness, Accountability & Transparency, PMLR 81, 134 - 148 Cao et al. (2018) "Deep Learning and Its Applications in Biomedicine" Genomics Proteomics Bioinformatics 16, 17 - 32 Yamashita et al. (2018) "Convolutional neural networks: an overview and application in radiology" Insights Imaging 9, 611 - 629 |
|
Mazo et al. (2018) "Transfer learning for classification of cardiovascular tissues in histological images" Comput. Methods Programs Biomed. 165, 69 - 76 Grapov et al. (2018) "Rise of Deep Learning for Genomic, Proteomic, and Metabolomic Data Integration in Precision Medicine" OMICS 22, 630 - 636 Heckmann et al. (2018) "Machine learning applied to enzyme turnover numbers reveals protein structural correlates and improves metabolic models" Nat. Commun. 9, 5252 |
|
Chan et al. (2019) "Advancing Drug Discovery via Artificial Intelligence" Trends Pharmacol. Sci. 40, 592 - 604 Zou et al. (2019) "A primer on deep learning in genomics" Nat. Genet. 51, 12 - 18 Eraslan et al. (2019) "Deep learning: new computational modelling techniques for genomics" Nat. Rev. Genet. 20, 389 - 403 Arute et al. (2019) "Quantum supremacy using a programmable superconducting processor" Nature 574, 505 - 510 |
|
Ham et al. (2019) "Deep learning for multi-year ENSO forecasts" Nature 573, 568 - 572 Alley et al. (2019) "Unified rational protein engineering with sequence-based deep representation learning" Nat. Methods doi: 10.1038/s41592-019-0598-1 AlQuraishi M. (2019) "AlphaFold at CASP13" Bioinformatics doi: 10.1093/bioinformatics/btz422 Gao et al. (2019) "DESTINI: A deep-learning approach to contact-driven protein structure prediction" Sci. Rep. 9, 3514 |
|
Strubell et al. (2019) "Energy and Policy Considerations for Deep Learning in NLP" arXiv:1906.02243 Wang et al. (2019) "Predicting Protein-Protein Interactions from Matrix-Based Protein Sequence Using Convolution Neural Network and Feature-Selective Rotation Forest" Sci. Rep. 9, 9848 Zhavoronkov et al. (2019) "Deep learning enables rapid identification of potent DDR1 kinase inhibitors" Nat. Biotechnol. 37, 1038 - 1040 Abu-Hassan et al. (2019) "Optimal solid state neurons" Nat. Commun. 10, 5309 Zou et al. (2019) "A primer on deep learning in genomics" Nat. Genet. 51, 12 - 18 |
|
Senior et al. (2020) "Improved protein structure prediction using potentials from deep learning" Nature 577, 706 - 710 Vandans et al. (2020) "Identifying knot types of polymer conformations by machine learning" Phys. Rev. E 101, 022502 Stokes et al. (2020) "A deep learning approach to antibiotic discovery" Cell 180, 688 - 702 |
|
Tunyasuvunakool et al. (2021) "Highly accurate protein structure prediction for the human proteome" Nature 596, 590 - 596 Walsh et al. (2021) "DOME: recommendations for supervised machine learning validation in biology" Nat. Methods 18, 1122 - 1127 Jumper et al. (2021) "Highly accurate protein structure prediction with AlphaFold" Nature 596, 583 - 589 Wu et al. (2021) "Strong Quantum Computational Advantage Using a Superconducting Quantum Processor" Phys. Rev. Lett. 127, 180501 Bepler & Berger (2021) "Learning the Protein Language: Evolution, Structure and Function" Cell Syst. 12, 654 - 669 |
|
Vaishnav et al. (2022) "The evolution, evolvability and engineering of gene regulatory DNA" Nature 603, 455 - 463 Schuman et al. (2022) "Opportunities for neuromorphic computing algorithms and applications" Nat. Comput. Science 2, 10 - 19 Chowdhury et al. (2022) "Single-sequence protein structure prediction using a language model and deep learning" Nat. Biotechnol. 40, 1617 - 1623 Pearce et al. (2022) "De Novo RNA Tertiary Structure Prediction at Atomic Resolution Using Geometric Potentials from Deep Learning" bioRxiv Sapoval et al. (2022) "Current progress and open challenges for applying deep learning across the biosciences" Nat. Commun. 13, 1728 |
|
Hekkelman et al. (2023) "AlphaFill: enriching AlphaFold models with ligands and cofactors" Nat. Meth. 20, 205 - 213 Santos-Junior et al. (2024) "Discovery of antimicrobial peptides in the global microbiome with machine learning" Cell S0092-8674(24)00522-1 |
|
![]()