| Eléments de base du langage Python et de quelques modules BioPython appliqués aux données biologiques |
|
|
1. Introduction 2. Quelques éléments de base 3. Les modules BioPython 4. L'interrogation d'une base de données - Les outils "E-Utils" de Entrez (NCBI) 5. L'interface Python Tutor |
6. Les expressions régulières ou motifs 7. Détail d'un script de recherche de motif - expression régulière 8. Scripts divers 9. Notions de base d'objet, de classe et d'attribut 10. Liens Internet et références bibliographiques |
|
Voir les thèmes abordés lors de la séance de travaux dirigés.
|
1. Introduction a. Pour exécuter un script Python :
b. L'environnement IDLE
|
|
Quelques différences Python 2 versus Python 3 |
||
| Caractéristique | Python 2 | Python 3 |
| Version de Python | Obsolète (dernière version 2.7) |
Python 3 a été publié en 2008 |
| Commande pour afficher la version de Python | python --version | python3 --version |
| Mot-clé "print" | instruction - pas de parenthèse [exemple : print "toto" ] | fonction - parenthèses [exemple : print ("toto") ] |
| Stockage des chaînes | en ASCII par défaut | en UNICODE par défaut |
| Division d'entiers | la division de 2 entiers donne un entier (exemple : 5/2 -> 2) | la division de 2 entiers donne un chiffre à virgule (exemple : 5/2 -> 2,5) |
| Exceptions | entourées de notations | entre parenthèses |
| (Rétro)compatibilité | Python 2 peut être porté vers Python 3 | pas compatible avec Python 2 |
| Voir le document différences Python V2 vs. Python V3 | ||
| 2. Quelques éléments de base | ||
Les mots-clés réservés en Python ne peuvent pas être utilisés comme nom de variable. and del from none true as elif global nonlocal try assert else if not while |
L'indentation : les blocs de code (fonctions, instructions, boucles, ...) sont délimités par une indentation (décalage du texte d'un ou plusieurs espace(s) ou tabulation(s)). L'indentation indique le début d'un bloc et la désindendation la fin de ce bloc. def fonction(n):
print 'n =', n
if n > 1:
return n * fonction(n - 1)
else:
print 'fin'
return 1
L'indentation nécessite 4 espaces selon la PEP8. Les espaces sont préferés aux tabulations. |
|
Le code situé après le caractère # (dièse) est igoré : il s'agit de commentaires . Les commentaires sont très importants pour bien décrire et comprendre un script. Exception : la commande #!/usr/bin/env python (voir ci-dessus) n'est pas interprétée comme un commentaire. |
Indentation - suite : tester les 2 scripts suivants et expliquer la différence de résultats : nucleotides = ['A','T','G','C']
for N in nucleotides:
if N == 'A':
print "la variable N"
print "a pour valeur", N
nucleotides = ['A','T','G','C']
for N in nucleotides:
if N == 'A':
print "la variable N"
print "a pour valeur", N |
|
Le symbole deux points : est également un délimiteur en Python. Il indique au langage qu'il doit attendre un bloc d'instructions : instruction 1: instruction2 instruction3 Les instructions for, while (boucles) et if (conditions) doivent se terminer par le symbole deux-points : liste = ['mot1','mot2','mot3'] for i in liste: print i #commentaire : indentation de cette instruction |
Interaction avec l'utilisateur
|
|
L'espace est le séparateur d'instructions :
|
Quelques messages d'erreurs Traceback (most recent call last): |
|
Lecture r : ouverture en lecture (READ). La méthode read() lit l'ensemble du contenu d'un fichier et renvoie une chaîne de caractères unique. |
Ecriture fichier = open("data.txt", "a")
fichier.write("Je m'appelle Toto")
fichier.close() |
|
Lecture - suite La méthode readline() (sans "s") :
La méthode readlines() (avec un "s") renvoie une liste avec toutes les lignes du fichier lu. Exemple : on applique la méthode "readlines()" sur l'objet "LireLesLignes" LireLesLignes = open('Fichier.fasta', mode = 'r')
LireLesLignes.readlines()
LireLesLignes.close() |
Exemple d'importation de bibliothèques import sys import re from Bio import Entrez
Source : Datacamp |
|
Créer une fonction def nom_fonction(parametre 1, parametre 2, parametre 3, ..., parametre n) : Bloc d'instructions La syntaxe :
Exemple (Python version 3.6) : def table_de_multiplication(max):
multiplicateur = input("choisissez un multiplicateur : ")
m = int(multiplicateur) #Evite : Type Error: < not supported between instances of int and str
max = input("choisissez un nombre maximum a multiplier : ")
n = int(max)
compteur = 0 # initialisation du compteur a 0
while compteur < n: # tant que compteur est strictement inferieur a la valeur de la variable max
print((compteur + 1),"*",(m), "=" ,(compteur + 1) * (m)) # attention aux parentheses pour print en 3.6
compteur += 1 # on incremente compteur de 1 a chaque tour de boucle
table_de_multiplication(max) # appel de la fonction
Execution du script a. Coller les lignes suivantes dans l'interpréteur Python (attention à l'indentation) def table_de_multiplication(max):
multiplicateur = input("choisissez un multiplicateur : ")
max = input("choisissez un nombre maximum a multiplier : ")
compteur = 0 # le compteur
while compteur < max: # Tant que compteur est strictement inferieur a la valeur de la variable max
print(compteur + 1),"*",(multiplicateur), "=" ,(compteur + 1) * (multiplicateur)
compteur += 1 # On incremente compteur de 1 a chaque tour de boucle
b. Taper 2 fois sur la touche "Entrer". On obtient : >>> def table_de_multiplication(max):
... multiplicateur = input("choisissez un multiplicateur : ")
... max = input("choisissez un nombre maximum a multiplier : ")
... compteur = 0 # le compteur
... while compteur < max: # Tant que compteur est strictement inferieur a la valeur de la variable max
... print(compteur + 1),"*",(multiplicateur), "=" ,(compteur + 1) * (multiplicateur)
... compteur += 1 # On incremente compteur de 1 a chaque tour de boucle
...
>>> |
|
BioPython fournit des fonctions et des procédures pour traiter et analyser des données biologiques avec le langage Python. Les scripts sont rassemblés sous forme de modules (et sous-modules) ou d'ensembles de script ("package") :
|
| Exemples de modules de la collection "Bio" pour la manipulation de séquences d'acides aminés et de nucléotides | ||||
| Nom du module (et lien internet) | But | Syntaxe pour l'import | Exemples de classes | Exemples de fonctions |
| Bio.AlignIO (Package AlignIO) | Multiple sequence alignment input/output as alignment objects. The Bio.AlignIO interface is deliberately very similar to Bio.SeqIO. | from Bio import AlignIO | ----- | ----- |
| Bio.Alphabet (Package Alphabet) | Alphabets used in Seq objects etc to declare sequence type and letters. | from Bio.Alphabet import IUPAC from Bio.Alphabet import generic_dna, generic_protein |
Alphabet, SingleLetterAlphabet, ProteinAlphabet, NucleotideAlphabet | _consensus_alphabet(alphabets) |
| Bio.Entrez (Package Entrez) | Provides code to access NCBI over the WWW. | from Bio import Entrez | ----- | esearch(db, term, **keywds) esummary(**keywds) einfo(**keywds) |
Exemple de script qui renvoie le titre et les auteurs de 2 publications dans Pubmed dont les identifiants sont "27297221" et "22615859" : from Bio import Entrez
Entrez.email = "imele@toto.mars"
requete = Entrez.einfo()
enregistrement = Entrez.read(requete)
requete.close()
requete = Entrez.esummary(db="pubmed", id="27297221,22615859", retmode="xml")
ListEnregistrements = Entrez.parse(requete)
for enregistrement in ListEnregistrements:
print "Titre de l'article :", (enregistrement['Title'])
print "Auteurs :", (enregistrement['AuthorList']) |
||||
| Bio.SearchIO (Package SearchIO) | Biopython interface for sequence search program outputs. | from Bio import SearchIO | ----- | parse, read, to_dict, index, index_db, write, convert |
| Bio.Seq (Module Seq) | Provide objects to represent biological sequences with alphabets. | from Bio.Seq import Seq | Seq, UnknownSeq, MutableSeq | transcribe(dna) translate(sequence, table='Standard', stop_symbol='*', to_stop=False, cds=False, gap=None) reverse_complement(sequence) complement(sequence) |
| Voir un complément sur Seq. | ||||
Bio.SeqIO (Package SeqIO) |
Sequence input/output as SeqRecord objects. | from Bio import SeqIO | ----- | convert(in_file, in_format, out_file, out_format, alphabet=None) parse(handle, format, alphabet=None) read(handle, format, alphabet=None) write(sequences, handle, format) |
|
Exemple de script avec un fichier "Fichier.fasta" disponible : from Bio import SeqIO
for record in SeqIO.parse("Fichier.fasta", "fasta"):
print(record.id)
Autre forme de ce script avec "with" et "handle": from Bio import SeqIO
with open("Fichier.fasta", "rU") as handle:
for record in SeqIO.parse(handle, "fasta"):
print(record.id) |
||||
| Bio.SeqUtils (Package SeqUtils) | Miscellaneous functions for dealing with sequences. | from Bio.SeqUtils import GC | ----- | GC(seq) nt_search(seq, subseq) six_frame_translations(seq, genetic_code=1) |
| Bio.motifs (Package motifs) | Tools for sequence motif analysis. | from Bio import motifs | Instances, Motif | create(instances, alphabet=None) parse(handle, format) read(handle, format) write(motifs, format) |
| Bio.pairwise2 (Module pairwise2) | Pairwise sequence alignment using a dynamic programming algorithm. | from Bio import pairwise2 | identity_match, dictionary_match, affine_penalty | _align(sequenceA, sequenceB, match_fn, gap_A_fn, gap_B_fn, ...) _make_score_matrix_generic(sequenceA, sequenceB, ...) _recover_alignments(sequenceA, sequenceB, ...) |
Exemple de script qui aligne 2 séquences de nucléotides : from Bio import pairwise2
from Bio.pairwise2 import format_alignment
alignments = pairwise2.align.globalxx("ACCGT", "ACG")
print(format_alignment(*alignments[0])) |
||||
4. L'interrogation d'une base de données - Les outils E-Utils de Entrez au NCBI Voir la syntaxe de tous les parametres des fonctions "esearch", "esummary", "efetch", "epost", "elink" et autres de Eutils - NCBI. L'URL de base est : https://eutils.ncbi.nlm.nih.gov/entrez/eutils/ Quelques exemples de parametres additionnels : motCle = raw_input ("Entrez votre mot cle : ") # "motCle" est renvoye dans la fonction "rechercheBiblio()"
def rechercheBiblio(motCle): # Definition de la fonction "rechercheBiblio()"
On genere l'URL suivante pour la requête au NCBI : https://eutils.ncbi.nlm.nih.gov/entrez/eutils/esearch.fcgi?db=pubmed&retmax=10&datetype=PDAT&retmode=xml&term=motCle Abréviations et syntaxe de quelques champs de fichiers GenBank ou GenPep : Liste des UIDs : Entrez Unique Identifiers for selected databases Voir un exemple de script Python 3.8 de recherche d'articles dans PubMed sur la base de mots clé. Il nécessite l'installation du module Bio de BioPython. |
5. L'interface WEB Python tutor Aller à "Python tutor". Coller le texte du script ci-dessous dans la fenêtre de l'interface : attention au respect de l'indentation de ce texte. Script Python version 3.6 : def denombre(sequence,nucleotide): # definition de la fonction denombre
compteur_nucleotide = 0 # compteur du nucleotide choisi
for nucleotide_lu in sequence:
if nucleotide_lu == nucleotide:
compteur_nucleotide += 1
return compteur_nucleotide
sequence = input("sequence a analyser : ")
nucleotide = input("nucleotide a denombrer : ") # input et non raw_input
n = denombre(sequence,nucleotide)
print ('%s apparait %s fois dans %s' % (nucleotide, n, sequence)) # doubler les parentheses
Cliquer sur "Visualize Execution".
Voir le script pour une séquence entrée transformée en majuscules et nettoyée avec une expression régulière. |
|
a. Syntaxe et signification de quelques expression régulières |
|
| Expression régulières | fonction |
| . |
tous les caractères sauf \n (retour chariot) pour désigner le caractère point ".", il faut le caractère d'échapement (anti-slash) et on écrit l'expression : \. |
| * | l'expression régulière qui précède est répétée 0 à n fois |
| + | l'expression régulière qui précède est répétée 1 à n fois => exemple : 0x[0-9ABCDEF]+ |
| (exp) | groupement des signes de l'expression régulière située dans la parenthèse (voir ci-dessous) Les parenthèses sont dites capturantes : l'expression régulière est placée dans une variable à laquelle on peut faire appel ailleurs. |
| exp_reg1 | exp_reg2 | l'opérateur | ("pipe") correspond à la recherche de l'expression régulière 1 ou de l'expression régulière 2 |
| [abc], [XYZ] | les lettres minuscules a ou b ou c - les lettres majuscules X ou Y ou Z |
| [a-z] [^a-z] |
les lettres minuscules de a à z = ensemble de l'alphabet tous les caractères sauf [a-z] |
| {n,m} {n,} {,m} |
l'expression régulière qui précède est retrouvée de au moins n fois à au plus m fois l'expression régulière qui précède est retrouvée au moins n fois l'expression régulière qui précède est retrouvée au plus m fois |
| \w \W |
tous les caractères alphanumériques (unicode), chiffres et _ (caractère souligné). C'est équivalent à la classe [a-zA-Z0-9_] tous les caractères non alphanumériques, c'est-à-dire les caractères non recherchés par \w. C'est équivalent à la classe [^a-zA-Z0-9_] |
| \s \S |
tous les caractères d'espacement (espace, tabulation, retour chariot, ...) - équivalent à la classe [ \t\n\r\f\v] tous les caractères qui ne sont pas caractères d'espacement (caractères non recherchés par \s) - équivalent à la classe [^ \t\n\r\f\v] |
| [0-9] ou \d [^0-9] ou \D |
tous les chiffres tous les caractères qui ne sont pas des chiffres, c'est-à-dire les caractères non recherchés par \d |
| ^ ou \A | caractère au début de la chaîne de caractères ou de la ligne |
| $ ou \Z | caractère à la fin de la chaîne de caractères ou de la ligne |
| \b \B |
caractère vide au début ou à la fin d'un mot caractère vide sauf au début ou à la fin d'un mot |
| \n | retour chariot |
| \t | tabulation |
| Syntaxes des expressions régulières et opérations en Python 3. | |
|
b. Compléments sur les métacaractères ou caractères spéciaux Les métacaractères sont : . ^ $ * + ? { } [ ] \ | ( ) Le métacaractère [ ] définit une classe de caractères. Le tiret (exemple [0-9]) traduit l'intervalle de la classe. Le métacaractère ^ a 2 significations tout à fait différentes : Le métacaractère $ : l'expression motif$ signifie que la chaîne de caractères recherchée se termine par le motif Le métacaractère ? : l'expression motif? signifie 0 ou 1 occurrence du motif |
|
| Quelques exemples | |
carottes? (pomme)+ |
Le quantificateur ? s'applique au caractère "s" et non au mot "carottes". Le quantificateur + s'applique à la sous-expression "(pomme)". |
| ^[a-z]*$ | La chaîne vide ou qui contient un ensemble de lettres minuscules. Elle se traduit par : commence par une lettre (^[a-z]) puis d'autre(s) (*) lettre(s) jusqu'à la fin ($). |
| -?[A-Z] | Une lettre majuscule (^[A-Z]) éventuellement (?) précédée (signe - écrit avant le métacaractère ?) du signe moins. |
| ^[ a-z]*$ | Attention à l'espace entre le crochet ouvrant et la lettre "a". Il s'agit donc d'une chaîne de caractères constituée de lettres minuscules séparées par un ou plusieurs espaces. |
| les métacaractères * et + renvoient la plus longue chaîne de caractères possible | Recherche du motif 1* dans la chaîne "xyz111E" qui renvoie une chaîne vide. |
| "et / ou" : + et | ("pipe") |
(xy)+ signifie xy ou xyxy ou xyxyxy ou ... x+y+ signifie n fois x suivis de n fois y (x|y)+ signifie une chaîne constituée de n fois x et/ou y |
Exemples de combinaisons d'expressions régulières
Afin d'en simplifier la lecture, on peut écrire une expression régulière complexe sur plusieurs lignes avec 3 apostrophes ('multiline string') en début et fin : expresionComplexe = re.compile(r'''( (\d{3}|\(\d{3}\))? (\s|-|\.)? \d{3} )''') |
|
Expression régulière correspondant à une chaîne de caractères d'une adresse mail Une adresse mail est écrite en général avec des caractères alphanumériques (éventuellement point ou tiret) puis le symbole @, puis le nom du domaine et enfin un suffixe (.fr, .com, .org ...). Exemple : nom@domaine.fr Expression régulière : ^[a-zA-Z0-9._%-+]+@[a-zA-Z0-9.-]+\.[a-zA-Z]{2, }$
|
|
Expression régulière correspondant à une chaîne de caractères d'une URL Exemple : https://www.UnSiteWeb.com Expression régulière : ^(https?)([A-Za-z0-9-]+\.)+[A-Za-z]{2,6}(:\d+)?(\[^\s]*)?$ |
|
Test sur les expressions régulières : aller à la page "Les expressions régulières en python" et voir le test "TRUE" / "FALSE". |
|
| c. Expressions régulières puissantes | ||
α. Recherche en avant texte(?=motif) : expression régulière qui vérifie que le texte est suivi du motif avant de sélectionner ce texte. C'est une recherche "positive en avant" ("positive lookahead"). texte(?!motif) : expression régulière qui vérifie que le texte n'est pas suivi du motif avant de sélectionner ce texte. C'est une recherche "négative en avant" ("negative lookahead"). Le texte et le motif peuvent être une expression complexe. Exemple : l'expression Isaac(?=Asimov) récupère "Isaac" seulement si il est suivi de "Asimov". |
β. Recherche en arrière (?<=motif)texte : expression régulière qui vérifie que le texte est précédé du motif avant de sélectionner ce texte. C'est une recherche "positive en arrière" ("positive lookbehind"). (?< !motif)texte : expression régulière qui vérifie que le texte n'est pas précédé du motif avant de sélectionner ce texte. C'est une recherche "négative en arrière" ("negative lookbehind"). Attention : le texte et le motif ne peuvent pas être une expression complexe. Exemple 1 : l'expression (?<!a)b reconnait un "b" non précédé d'un "a". Cette expression ne reconnait donc pas le "b" de "table" mais reconnait le "b" (et seulement le "b") de "banane". Exemple 2 :
import re
m = re.search("(?<=abc)def", "abcdef")
m.group(0)
'def' |
|
| Voir un développement. | ||
d. Quelques méthodes liées aux expressions régulières ou motifs
|
Les méthodes "match()" et "search()" renvoient "None" si aucune correspondance n'est trouvée. Si une correspondance est trouvée, elles renvoient une instance "MatchObject" qui contient les informations sur la correspondance : son début et sa fin, la sous-chaine avec laquelle elle correspond,... |
|
|
La méthode "match()" recherche une correspondance au début de la chaîne de caractères. Attention à la syntaxe (apostrophe, virgule de séparation, parenthèses). |
correspondance = re.match('motif','chaîne de caractères dans laquelle rechercher le motif') |
|
La méthode "search()" recherche une correspondance dans toute la chaîne de caractères et/ou dans la première ligne du texte. L'option "re.MULTILINE" (= re.M) permet de chercher dans toutes les lignes. |
correspondance = re.search('motif','chaîne de caractères dans laquelle rechercher le motif') Exemple : |
La méthode "sub()" remplace toutes les occurrences du motif par un autre motif. La méthode "sub()" renvoie la chaîne de caractères modifiée. |
correspondance = re.sub('motif','motif de remplacement','chaîne de caractères dans laquelle rechercher l'expression') Exemples : |
La méthode "compile()" Il peut être avantageux en terme de mémoire et de temps de conserver une version compilée du motif. Par exemple, déclaration et compilation une seule fois au démarrage d'un serveur => ces opérations ne sont pas refaites à chaque requête. La méthode "compile()" renvoie un objet de type expression régulière ("<_sre.SRE_Pattern object at 0x1055fcac0>") qui peut ètre utilisée avec les méthodes "search()" ou "findall()". |
Exemple 1 : Exemple 2 : |
Application recherche 'positive et négative en avant' recherche 'positive et négative en arrière' |
correspondance = re.compile(r'(?!pique)e') correspondance = re.compile(r'(?<=pique)e') correspondance = re.compile(r'(?<!pique)é') |
La méthode "findall()"renvoie les éléments qui correspondent sous forme d'une liste.
La méthode "findall()" peut-être appliquée à un fichier. |
Exemple 1 : Exemple 2 : |
|
La méthode "split()"découpe - sépare la chaîne de caractères selon le motif.
Elle renvoie les éléments qui correspondent sous forme d'une liste. |
correspondance = re.split('motif','chaîne de caractères dans laquelle rechercher l'expression') Exemple 1 : Exemple 2 : |
| Voir l'ensemble des méthodes à base d'expressions régulières (ou rationnelles) - Python version 3.6. | |
7. Détail d'un script de recherche de motif - expression régulière
|
| # !/usr/bin/env python # -*-coding:Utf-8 -* import sys import re from Bio import SeqIO |
# Le systeme SeqIO retourne des objects de type "SeqRecord" |
| def LectureFichier(FichierTraite): try : OuvertureFichier = open(FichierTraite, 'r') except IOError : print ("Erreur d'ouverture de fichier"),FichierTraite sys.exit() |
# 1ere partie : FONCTION lecture de fichier # Test ouverture du fichier et instruction de sortie si erreur # la variable "FichierTraite" designe n'importe quel fichier que l'on desire ouvrir # Attention à l'indentation (espace) |
| indice = 0 for ChaqueSequence in SeqIO.parse(FichierTraite, 'fasta'): indice = indice+1 print ("Sequence numero"), indice print(ChaqueSequence.format("fasta")) |
# 2eme partie : "parsing" des differents items qui commencent par ">" du fichier a lire # Si fichier GenBank (par exemple) => remplacer ".fa" par ".gbk" et "fasta" par "genbank" #.format() est une methode qui convertit l'objet "SeqRecord" en une chaîne en utilisant l'une des options de sortie de Bio.SeqIO # Attention à l'indentation (espace) |
| PremiereLigneFichier = OuvertureFichier.readline().strip() | # 3eme partie : Decoupage des lignes du fichier lu # la methode "readline()" lit une ligne a la fois => en l'occurence la 1ere ligne : ">Texte informatif" # cette methode n'est appelee qu'une fois donc seule la 1ere ligne est traitee / # la methode readline() renvoie une chaîne DE CARACTERES # la methode "strip()" enleve "\n" de fin de ligne |
| Sequence = '' | # initialise la variable chaîne de caractere "sequence" qui est vide au depart |
| for ligne in OuvertureFichier.readlines(): | # methode "readlines()" : attention au "s" cette fois / cette methode lit toutes les lignes ("for") # a partir de la 2eme ligne puisque readline() precedent a place le curseur sur la 2eme ligne # la methode readlines() renvoie une LISTE |
| ligne.strip() Sequence = Sequence + ligne.strip() return(PremiereLigneFichier, Sequence) OuvertureFichier.close() |
# la methode "strip()" enleve "\n" de fin de chaque ligne en cours # concatene la ligne en cours avec la precedente => la variable sequence ajoute chaque nouvelle ligne lue # renvoie 2 variables : la variable PremiereLigneFichier et la variable Sequence # Attention a l'indentation sinon "NameError: name 'OuvertureFichier' is not defined |
| def RechercheMotif(Motif, ChainAtraiter): | # FONCTION recherche du motif # "Motif" est le nom generique de la variable specifique passee en 1er parametre lors de l'appel de la fonction "RechercheMotif" # "ChainAtraiter" est le nom generique de la variable specifique passee en 2e parametre lors de l'appel de la fonction "RechercheMotif" (dans le cas present la variable "sequence") |
| MotifTransforme = re.compile(Motif) MotifTrouve = re.finditer(MotifTransforme, ChainAtraiter) |
# la methode "compile" : transforme en veritable expression reguliere # la methode "finditer" : recherche toutes les occurences du motif considere de gauche a droite et enregistre les positions de ce motif # ces positions sont renvoyees par la suite avec les methodes "start()" et "end()" ci-dessous |
| for position in MotifTrouve: print position.start(),"-",position.end()," : ", position.group() return MotifTrouve |
# Exemple de positions de debut et de fin d'un motif trouve => 2 - 6 : HLGH et la methode "group()" renvoie le motif trouve # attention : "print position" est indente 2 fois # attention : indenter le "return" |
| FichierTraite = input("Nom du fichier de sequence : ") (PremiereLigneFichier, Sequence) = LectureFichier(FichierTraite) |
# Entree des donnes a traiter : fichier a lire et motif recherche |
| print ("Resultat intermediaire") print ("Variable Premiere ligne du fichier : "), PremiereLigneFichier print ("Variable Sequence : "), Sequence print ("Longueur de la sequence : "), len(Sequence) |
# Divers affichages |
| ExprREGUL = input("Expression reguliere / exemple : GA[ATGC]{3}AC : ") | # Saisir une expression reguliere au clavier |
| print ("Resultat final") ResExpRegul = RechercheMotif(ExprREGUL, Sequence) |
# Affichage des resultats # prend comme parametre de la fonction "RechercheMotif" : Motif = ExprREGUL et ChainAtraiter = Sequence |
| But des scripts | scripts (Python v. 2.7) | via une page web |
| Afficher les identités entre 2 séquences | Affichage identite | ----- |
| Utilisation de "def" et "while" / Données saisies par l'utilisateur | Table multiplication | ----- |
| Dénombrement des nucléotides dans une séquence et calcul du Tm Entrer une sequence et la nettoyer / ouvrir et lire un fichier / liste et dictionnaire / affichages Description de l'utilisation de Python tutor |
||
|
Recherche d'articles dans PubMed - NCBI (via Entrez et les outils Eutils) sur la base de mots clés Module "from Bio import Entrez" / module "urllib.request" |
Entrez (NCBI) | Recherche PUBMED |
| Utilisation de "def" et "while" - Données saisies par l'utilisateur Module "re" / module "from Bio import SeqIO" |
Reg_Exp Motif Fichier test.fa |
----- |
| Calcul de l'hydrophobicité moyenne d'un peptide - dictionnaire et liste - récurrence | Hydrophobicité | ----- |
| Calcul de l'hydrophobicité moyenne d'un peptide - fichier de valeurs externe Entrer une sequence et la nettoyer / liste et dictionnaire/ affichage du décours du calcul/ écrire et lire un fichier |
Hydrophobicité Fichier hydrophobicite.txt |
Hydrophobicité moyenne |
|
Profil d'hydrophobicité affiché avec une fenêtre de taille n = 7 ou variable. Bibliothèques SeqIO, pylab et numpy. Voir le cours : "Bionformatique : analyse et prédiction des structures des protéines". |
Profil hydrophobicité | |
| Interrogation de la base de données NCBI - Récupération des informations GenPept : outils Eutils Classe et objet / liste et dictionnaire Bibliothèques BioPython, pylab et numpy. Voir le cours : "Relation structure - fonction des protéines". |
----- |
Propriétés physico-chimiques d'une protéine |
| Structure des protéines | scripts (Python v. 2.7) | via une page web |
| Génération d'une URL (module "urllib.request") - Interrogation de la base de données PDB Recherche de la position des cystéines impliquées dans un pont disulfure Lecture - écriture dans un fichier |
Positions Ponts Python 2 Positions Ponts Python 3 |
Ponts disulfure PDB |
|
Interrogation de la base de données PDB (structure des protéines) Calcul du barycentre des carbones alpha des acides aminés |
Barycentre | Calcul du barycentre |
|
Classe et objet - bibliothèques BioPython, pylab et numpy. Voir le cours : "Les acides aminés et la structure primaire des protéines". |
----- |
Diagramme de Ramachandran |
|
Interrogation de la base de données PDB (structure des protéines) Calcul des distances entre les carbones alpha de 2 chaînes polypeptidiques Calcul et représentation des aires de contacts entre 2 chaînes polypeptidiques Module "Bio.PDB.PDBParser" |
Distance - contacts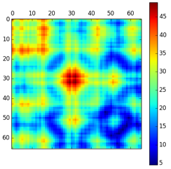 |
|
|
Interrogation de la base de données Pfam (domaines des protéines) Récupération d'un fichier HMM ("Hidden Markov models") |
Recherche d'un profil HMM | |
|
Interrogation de la base de données PDB Calcul du RMSD entre 2 structures de protéines : module "Bio.PDB.Superimposer()" |
----- | Superposition structures |
Enzymologie (voir le cours "Effets de paramètres physico-chimiques sur l'activité enzymatique") |
||
Bibliothèques MatplotLib et numpy Variation de la vitesse d'une réaction enzymatique en fonction de la température et du temps d'incubation. Graphique ci-contre tracé avec les valeurs : ΔG‡cat = 50 kJ.mol-1 ; ΔG‡inact = 96 kJ.mol-1 ; [E]0 = 10-5 M. |
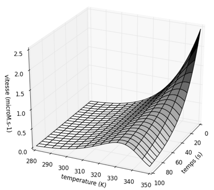 |
|
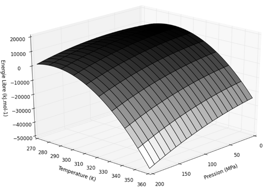
Variation d'énergie libre de Gibbs du processus de dénaturation d'une enzyme en fonction de la température et de la pression. Graphique ci-contre tracé avec les valeurs : Δα = - 1 mL.K-1.mol-1; Δβ = 0,65 mL.MPa-1.mol-1; ΔCp = 5,7 kJ.K-1.mol-1; ΔV0 = -8 mL.mol-1; ΔS0 = 0,28 kJ.mol-1; ΔG0 = 10,8 kJ.mol-1; T0 = 310 K; P0 = 0,1 Mpa. |
 |
|
| CinEnz (non opérationnel) KEGG (non opérationnel) |
||
9. Notions de base d'objet, de classe et d'attribut Définition de Cordeau, B. & Pointal, L. dans l'ouvrage "Python 3" (2017) : Les objets : le langage Python est un langage orienté objet :
Les attributs : ce sont les variables qui caractérisent un objet. Les classes : selon la PEP 8 ("Python Enhancement Proposals") de Python, il est préférable d'utiliser la convention "Camel Case" pour le choix du nom d'une classe :
Pour définir une classe, on emploie le mot-clé "class" selon la syntaxe : "class NomDeLaClasse:" (les 2 points font partie de la syntaxe). Une classe définit des attributs et des méthodes. Exemple : la classe « Personne » permet de créer des objets qui sont des « individus ».
Une autre classe « Oiseau » peut définir la méthode « voler( ) ».
Les instances de classe : un objet est une instance de classe. Chaque objet a des attributs qui lui sont propres. Il peut exister plusieurs objets pour une même classe, c'est-à-dire plusieurs instances de la même classe. Exemple, dans la classe Personne, il existe des individus qui sont des instances distinctes :
|
| 10. Liens Internet et références bibliographiques | |
The Python Tutorial : différentes versions sont décrites Informations sur les modules Python "Un petit tuteur sur les Expressions Régulières" - G. Hunault |
|
"Python 3" (2017) Cordeau, B. & Pointal, L. - ISBN 978-2-10-076636-9 - Editions DUNOD "Informatique avec Python" (2018) Beury J.N. - ISBN 978-2-10-076901-8 - Editions DUNOD "Introduction au machine learning" (2018) Azencott C.A. - ISBN 978-2-10-078080-8 - Editions DUNOD "Programmation en Python pour les sciences de la vie" (2019) Fuchs P. & Poulain P. - EAN 978-2-10-079602-1 - Editions DUNOD |
|
|
PeptideBuilder : bibliothèque pour générer des peptides modèles |
|
|
TP - Programmation Python pour la BIOInfo Expressions régulières (P. Vanier) |
|
|
Ebrahim et al. (2013) "COBRApy: COnstraints-Based Reconstruction and Analysis for Python" BMC Syst. Biol. 7, 74 La bioinformatique avec Biopython Olivier Dameron, Gregory Farrant Shajii et al. (2021) "A Python-based programming language for high-performance computational genomics" Nat. Biotechnol. 39, 1062 - 1064 |
|