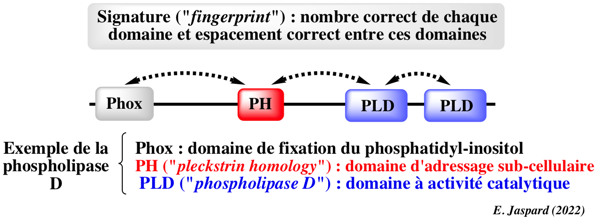
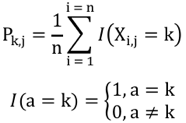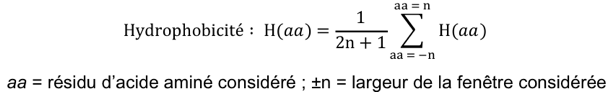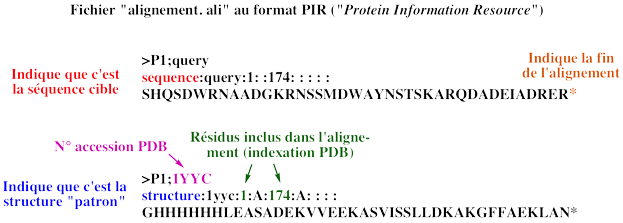a. Les scores de distances inter-atomiques entre des structures comparées
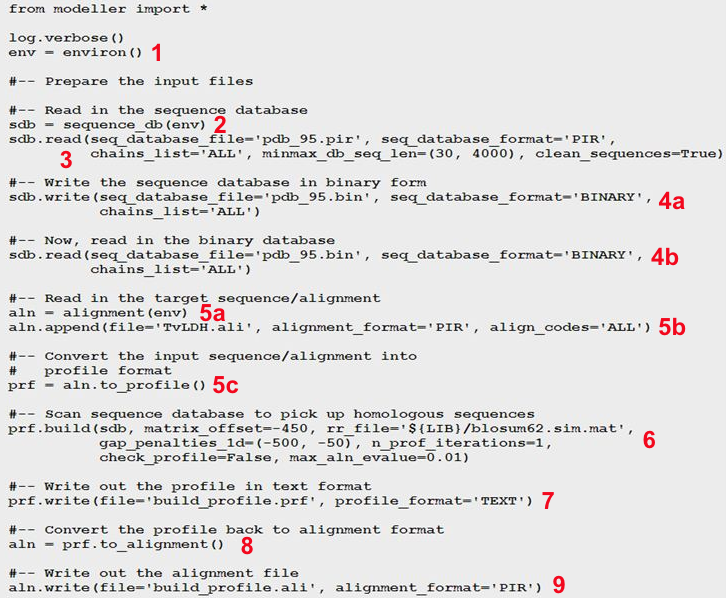
La modélisation par homologie de structures nécessite de superposer les structures des protéines qui sont comparées pour établir la correspondance spatiale entre les acides aminés équivalents dans ces structures.
Le score RMSD et le score RMSD local
Pour mesurer ces distances inter-atomiques on utilise fréquemment une grandeur appelée écart quadratique moyen (RMSD - "Root Mean Square Deviation", en Å) entre les coordonnées spatiales des atomes des acides aminés appariés.
Par exemple, si on considère n atomes de 2 structures, on compare les coordonnées (xi, yi, zi) d'un atome i de l'une de ces structures aux coordonnées (x'i, y'i, z'i) d'un atome i apparié de l'autre structure :
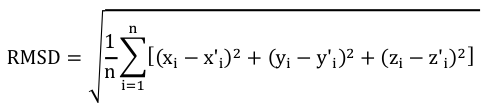
On superpose ainsi les structures en minimisant la valeur de RMSD.
- structures identiques : RMSD = 0
- structures similaires : RMSD = 1 - 3 Å
- structures éloignées : RMSD > 3 Å
Limites du score RMSD
- Il faut que les 2 structures soient dans le même système de repères orthonormés.
- Cette méthode attribue un poids statistique équivalent à tous les atomes (carbone α et atomes des chaînes latérales).
- Il faut préciser la liste des atomes à comparer : c'est un problème si les protéines n'ont pas des séquences de longueurs identiques.
Pour effectuer un choix pertinent des acides aminés à comparer, on calcule un RMSD local avec les carbones α des acides aminés inclus dans une fenêtre de longueur L+1 :
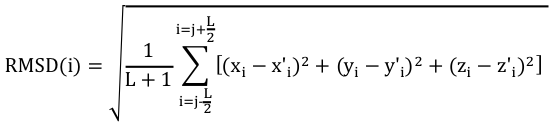
Le score GDT
Le score GDT ("Global Distance Test") est aussi une mesure de la similarité entre 2 structures protéiques qui ont des structures 3D différentes.
Ce score est calculé sur la base du plus grand ensemble de carbones α des résidus d'acides aminés de la structure experimentale dont la position dans l'espace correspond (selon un seuil donné : 1 Å, 2 Å, 4 Å et 8 Å) à celles des résidus d'acides aminés de la structure prise pour modèle.
Les scores GDT sont les principaux critères d'évaluation des résultats de prédiction issus de l'expérience CASP ("Critical Assessment of Structure Prediction") :
- CASP est une expérience à grande échelle de la communauté scientifique qui travaille sur la prédiction de structure.
- CASP évalue et améliore les différentes techniques de modélisation de structures de protéines.
Le score TM
Le score TM ("Template Modeling score") est également une mesure de la similarité entre 2 structures protéiques qui ont des structures 3D différentes.
Le score TM est une mesure plus précise de la qualité des structures protéiques que le score RMSD et le score GDT souvent utilisés. Dans le calcul du score TM, les distances faibles ont un poids plus élevé que les distances fortes : ce score est donc insensible aux erreurs de modélisation locale.
- Un score TM > 0. 5 indique une topologie correcte du modèle calculé : les protéines comparées ont un repliement similaire.
- Un scoreTM < 0. 17 correspond à des protéines non apparentées, choisies au hasard.
- Ces seuils de score ne dépendent pas de la longueur de la protéine.
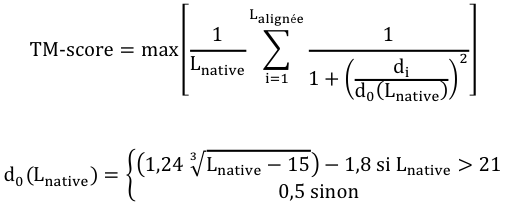
- max : la somme doit être maximisée (matrice de superposition optimale).
- Lnative est la longueur de la protéine native (la structure de référence avec laquelle s'effectue la comparaison) et Laligné est la longueur de la région d'acides aminés qui est alignée.
- di est la distance entre la ième paire de résidus d'acides aminés alignés entre les 2 structures. Cette distance dépend de la matrice de superposition.
- d0(Lnative) est une échelle de distance qui normalise les différences de distances.
Exemples de comparaisons et de scores de comparaisons
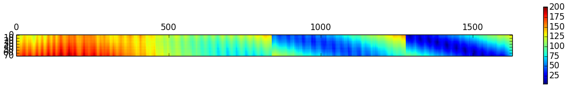
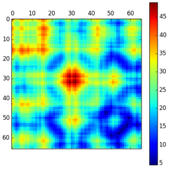
Figure ci-dessous : superpositions des structures de 2 protéines avec différents algorithmes (CE - "Combinatorial Extension of the optimal path", SAL, DALI et TM-align).
- Code PDB 1ATZ_A (184 résidus d'acides aminés) : domaine A3 du facteur Von Willebrand humain (fixation du collagène).
- Code PDB 1AUO_A (218 résidus d'acides aminés) : carboxylestérase.
- Ces 2 protéines ont 16% d'identité de séquence et elle adoptent une topologie en sandwich αβα.
- Les rubans épais et minces indiquent les résidus d'acides aminés alignés de 1ATZ_A et 1AUO_A, respectivement.
L est le nombre de résidus d'acides aminés alignés. Les scores RMSD et TM sont indiqués.
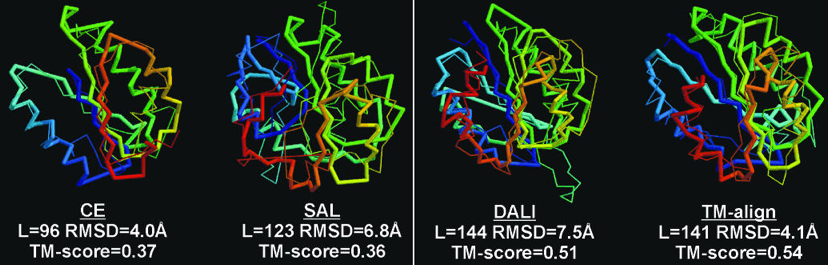
Source : Zhang & Skolnick (2005)
Le score lDDT
Le score lDDT ("local Distance Difference Test") mesure la façon dont l'environnement dans une structure référence est reproduit dans une structure modèle. Ce score est calculé sur toutes les paires d'atomes dans la structure de référence à une distance inférieure à un seuil prédéfini (appelé rayon d'inclusion) et n'appartenant pas au même résidu d'acide aminé.
Ces paires d'atomes définissent un ensemble de distances locales L :
- Une distance est conservée dans le modèle M si elle est (selon un seuil de tolérance) la même que la distance correspondante dans l'ensemble L.
- Si un ou les deux atomes définissant une distance dans l'ensemble L ne sont pas présents dans le modèle M, la distance est non conservée.
Pour un seuil donné, la fraction des distances conservées est calculée. Le score IDDT est la moyenne de 4 fractions calculées en utilisant des seuils identiques à ceux utilisés pour calculer le score GDT (voir ci-dessus).
Voir Mariani et al. (2013).
Divers
- Il existe un très grand nombre d'algorithmes et de programmes de superposition 2 à 2 ou multiples de structures de protéines ou d'autres molécules comme les ARN. Voir un très grand nombre de programmes de superposition de structures.
- SupeRNAlign : service WEB et programme en Python de superposition de structures d'ARN homologues.
|