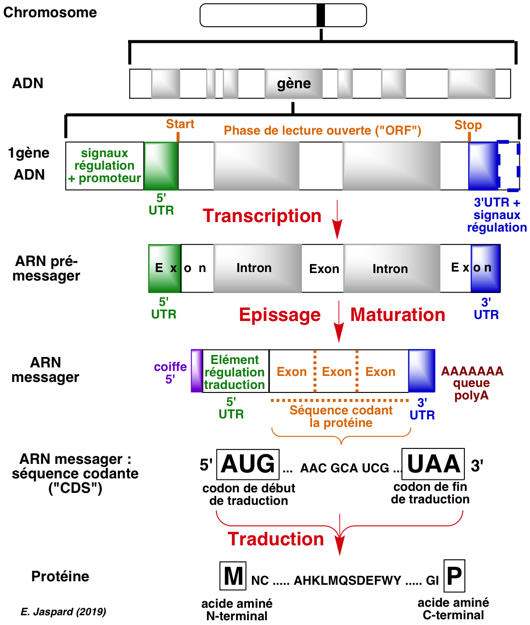
Figure ci-contre : représentation schématique du processus de transcription puis de traduction et de la structure des molécules issues de ces processus.
Cette étude bioinformatique des toxines de serpents parcourt ces protéines jusqu'à la structure 3D en focalisant sur les cystéines impliquées dans des ponts disulfure.
ExPASy : ensemble d'applications pour l'analyse de séquences peptidiques => sélectionner "Proteins & Proteomes" dans le menu de gauche.
EMBOSS seqret : application pour la conversion de formats de fichiers.

{kind=link}