| Quelques formats de fichiers dans les bases de données |
| Tweet |
|
Remarques préalables Le format des fichiers bioinformatiques est intimement lié aux logiciels - programmes qui les utilisent et aux bases de données qui les hébergent; La standardisation de certains formats (plus grande communicabilité / échange de fichiers plus pratique) et la disparition de certains logiciels ou certaines bases de données rendent caduques certains formats. A l'inverse, l'avènement de nouvelles technologies et/ou programmes est lié à la création de nouveaux formats de fichiers bioinformatiques. Par exemple :
Malré tout, le très grand nombre de formats (voir tableau ci-dessous) est un handicap pour un échange efficace et une utilisation optimale des données biologiques en bioinformatique. Une plus grande harmonisation est nécessaire. |
| Exemples de formats de fichiers bioinformatiques | |||
| Nom du format | Valeur | Nom du format | Valeur |
| EMBL entry format | embl | Plain text | text |
| GCG sequence format | gcg | Fitch program format | fitch |
| EMBL new entry format | emblnew | GCG MSF (multiple sequence file) file format | msf |
| Swissprot entry format | swiss | Clustalw multiple alignment format | clustal |
| Swissprot entry format (swold) | swold | NCBI ASN.1 format | asn1 |
| Swissprot entry format (swissnew) | swissnew | Hennig86 output format | hennig86 |
| FASTA format | fasta | Mega interleaved output format | mega |
| NCBI fasta format with NCBI-style IDs | ncbi | Nexus/paup interleaved format | nexus |
| NCBI fasta format with NCBI-style IDs using GI number | gifasta | Jackknifer output interleaved format | jackknifer |
| NBRF/PIR entry format | nbrf | Treecon output format | treecon |
| Genbank entry format | genbank | FASTQ short read format with phred quality | fastq |
| Genbank/DDBJ entry format (alias) | ddbj | FASTQ Illumina 1.3 short read format | fastq-illumina |
| Genpept entry format | genpept | FASTQ Solexa/Illumina 1.0 short read format | fastq-solexa |
| Refseq entry format | refseq | Sequence alignment/map (SAM) format | sam |
| GFF2 feature file with sequence in the header | gff2 | Binary sequence alignment/map (BAM) format | bam |
| Old staden package sequence format | staden | Debugging trace of full internal data content | debug |
| Voir "File formats used in bioinformatics". | |||
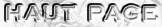
Format FASTA C'est sans doute le format de fichier le plus répandu car trés simple et l'un des plus pratiques. >gi|22777494|dbj|BAC13766.1| glutamate dehydrogenase [Oceanobacillus iheyensis] MVADKAADSSNVNQENMDVLNTTQTIIKSALDKLGYPEEVFELLKEPMRILTVRIPVRMDDGNVKVFTGY 1. Un fichier au format FASTA peut contenir plusieurs séquences. Chaque séquence (écrite sous forme de lignes de 80 caractères maximum), est précédée d'une ligne de titre (nom, définition ...) qui doit commencer par le caractère ">". 2. gi|22777494 : l'identifiant gi ("GenInfo Identifier") est le numéro d'identification d'une séquence (acides aminés ou nucléotides). Si une séquence est modifiée, un nouveau numéro de GI est attribué. Voir ci-dessous. 3. dbj|BAC13766.1| : un enregistrement peut exister dans différentes bases de données et peut avoir plusieurs identifiants. Le tableau ci-dessous donne l'explication du nom de la base de données et la syntaxe des identifiants. Dans cet exemple, l'enregistrement existe dans la base de données du Japon DDBJ sous l'identifiant dbj|BAC13766.1. 4. dbj|BAC13766.1| : les identifiants de séquences dans une base de données évoluent en parallèle avec la version du fichier. Dans cet exemple, le «.1» indique que la séquence a été modifiée une fois et qu'un nouveau fichier a été déposé. 5. glutamate dehydrogenase [Oceanobacillus iheyensis] : description de la séquence. Dans cet exemple, glutamate déshydrogénase est le nom de la protéine et Oceanobacillus iheyensis celui de l'organisme à partir duquel elle a été obtenue. |
| Base de données | Syntaxe de l'identifiant | Base de données | Syntaxe de l'identifiant |
| GenBank | gb|accession|locus | NCBI Reference Sequence | ref|accession|locus |
| EMBL Data Library | emb|accession|locus | Protein Research Foundation | prf||name |
| DDBJ (DNA Database of Japan) | dbj|accession|locus | Local Sequence identifier | lcl|identifier |
| NBRF PIR | pir||entry | GenInfo Backbone Id | bbs|number |
| UniProtKB/Swiss-Prot | sp|accession|entry name | General database identifier | gnl|database|identifier |
| PDB (Protein Data Bank) | pdb|entry|chain | Patents | pat|country|number |
Exemple du programme d'alignement multiple MUSCLE
L'en-tête de FASTA : un N° d'accession de la séquence avec la version et le titre de l'enregistrement pour les N° d'accession gérés par l'INSDC ("International Sequence Database Collaboration") et le projet RefSeq. Il semble que le NCBI continuera à fournir des informations sur la base de données source d'un fichier dans l'en-tête de FASTA, en particulier pour SwissProt, PDB ("Protein Data Bank"), PIR ("Protein Information Resource") et les séquences brevetées. |
|
Fichiers issus des nouvelles technologies de séquençage à très haut débit Voir les techniques de séquençage. 3 types de formats de fichiers sont couramment utilisés :
Description du format FASTQ C'est un format basé sur du texte pour stocker à la fois une séquence biologique (séquence nucléotidique habituellement) et ses scores de qualité. Fichier ci-dessous : première ligne = nom de la séquence après le symbole @ (et, éventuellement, la description) / deuxième ligne = la séquence / quatrième ligne = scores de qualité codés sous forme de lettres. @sequence 1 |
|
Format et base de données RefSeq RefSeq ("The Reference Sequence database") : base de données de séquences annotées (ADN, ARN, protéines) dites de référence. Elle est est construite par le NCBI. Contrairement à GenBank, elle ne fournit qu'un seul enregistrement (séquences non redondantes) pour chaque molécule biologique naturelle (ADN, ARN ou protéine) pour les principaux organismes (virus, bactéries, eucaryotes - # 113.000 organismes en octobre 2021). |
Figure ci-dessous : suite logicielle ("pipeline") de construction d'un fichier RefSeq.
Chaque enregistrement de RefSeq représente une synthèse (par une personne ou un groupe - "synthesizing editors") de l'ensemble des informations générées et soumis par la communuté scientifique. L'ensemble des données RefSeq est curé par des groupes de collaborateurs et par les curateurs du NCBI. |
| Signification des préfixes des N° d'accession des fichiers de la base de données RefSeq | ||
| préfixe du N° d'accession | type de molécule | commentaire |
| AC_ | Génomes | Complete genomic molecule, usually alternate assembly |
| NC_ | Complete genomic molecule, usually reference assembly | |
| NG_ | Incomplete genomic region | |
| NT_ | Contig or scaffold, clone-based or WGS ("Whole Genome Shotgun sequence data") | |
| NW_ | Contig or scaffold, primarily WGS | |
| NS_ | Environmental sequence | |
| NZ_ | Unfinished WGS | |
| NM_ | ARN messagers | ------- |
| NR_ | ARN | ------- |
| XM_ | ARN messagers | Predicted model (computed) |
| XR_ | ARN | Predicted model (computed) |
| AP_ | Protéines | Annotated on AC_ alternate assembly |
| NP_ | Associated with an NM_ or NC_ accession | |
| YP_ | (computed) | |
| XP_ | Predicted model, associated with an XM_ accession (computed) | |
| ZP_ | Predicted model, annotated on NZ_ genomic records (computed) | |
Exemples d'outils de conversion de formats "EMBOSS Seqret" : programme de l'EBI. Il permet le reformatage général des séquences. Le programe "GenBank to FASTA" prend en entrée un fichier GenBank et retourne la séquence d'ADN entière au format FASTA. A utiliser quand on veut supprimer les informations non ADN à partir d'un fichier GenBank. Divers |
|
Format EMBL Chaque entrée de la base de données EMBL est composée de lignes qui commencent par un code à deux caractères (champs) suivi par des informations. ID 1YYCA STANDARD; PRT; 174 AA |
Format Stockholm C'est le format des entrées de la base de données Pfam (collection de familles de domaines des protéines). C'est un format d'alignement multiples de séquences. Exemple pour le domaine CBS ("Cystathionine-β-synthase") : # STOCKHOLM 1.0
#=GF ID CBS
#=GF AC PF00571
#=GF DE CBS domain
#=GF AU Bateman A
#=GF CC CBS domains are small intracellular modules mostly found in 2 or four copies within a protein.
#=GF SQ 5
#=GS O31698/18-71 AC O31698
#=GS O31698/88-139 OS Bacillus subtilis
O83071/192-246 MTCRAQLIAVPRASSLAEAIACAQKMRVSRVPVYERS
#=GR O83071/192-246 SA 9998877564535242525515252536463774777
O83071/259-312 MQHVSAPVFVFECTRLAYVQHKLRAHSRAVAIVLDEY
#=GR O83071/259-312 SS CCCCCHHHHHHHHHHHHHEEEEEEEEEEEEEEEEEEE
O31698/18-71 MIEADKVAHVQVGNNLEHALLVLTKTGYTAIPVLDPS
#=GR O31698/18-71 SS CCCHHHHHHHHHHHHHHHEEEEEEEEEEEEEEEEHHH
O31698/88-139 EVMLTDIPRLHINDPIMKGFGMVINN..GFVCVENDE
#=GR O31698/88-139 SS CCCCCCCHHHHHHHHHHHHEEEEEEEEEEEEEEEEEH
#=GC SS_cons CCCCCHHHHHHHHHHHHHEEEEEEEEEEEEEEEEEEH
O31699/88-139 EVMLTDIPRLHINDPIMKGFGMVINN..GFVCVENDE
#=GR O31699/88-139 AS ________________*____________________
#=GR O31699/88-139 IN ____________1____________2______0____
//
Composition minimale : Un en-tête qui indique le format et la version de l'identifiant : # STOCKHOLM 1.0 |
| Format de la base de données PROSITE |
| Ligne | Exemple | Remarques |
| identification (ID) | ZF_FCS; MATRIX | 2 à 21 caractères alphanumériques majuscules |
| numéro d'accession (AC) | PS51024 | format : PSnnnnn avec PS = PROSITE et nnnnn = nombre à 5 chiffres |
| date (DT) | 01-NOV-2004 CREATED | la date de dépôt de l'entrée et des dernières modifications de l'entrée |
| description (DE) | Zinc finger FCS-type profile. | toujours la 4ème ligne - informations sur le contenu de l'entrée |
| pattern (PA) | F-[GSTV]-P-R-L-[G>] | syntaxe des expressions régulières |
| matrice / profil (MA) | MA /GENERAL_SPEC: ALPHABET='ABCDEFGHIKLMNPQRSTVWYZ'; LENGTH=36; MA /DISJOINT: DEFINITION=PROTECT; N1=4; N2=33; MA /NORMALIZATION: MODE=-1; FUNCTION=LINEAR; R1=1506.6335449; R2=1.4524887; PRIORITY=1; TEXT='Heuristic 5.0%'; MA /CUT_OFF: LEVEL=0; SCORE=733; H_SCORE=2571; N_SCORE=9.0; MODE=1; TEXT='!'; MA /DEFAULT: M0=-9; D=-20; I=-20; B1=-100; E1=-100; MI=-105; MD=-105; IM=-105; DM=-105; MA /I: B1=0; BI=-105; BD=-105; MA /M: SY='P'; M=-8,-7,-29,-2,12,-25,-16,-9,-24,4,-26,-18,-5,24,3,8,3,-4,-23,-30,-21,5; ... |
|
Autres lignes : modifications post-traductionnelles (PP) / commentaires (CC) / références UniProtKB-Swiss-Prot (DR) / références PDB (3D) / règle ProRule associée (PR) |
||
| Exemples | PS50011 | Prorule PRU00159 |
La syntaxe pour la description d'un motif structural ou signature ("pattern") de la base de données PROSITE est la suivante :
Exemples de motifs PROSITE : D-x-[DNS]-{ILVFYW}-[DENSTG]-[DNQGHRK]-{GP}-[LIVMC]-[DENQSTAGC]-x(2) |
Format ClustalW- suffixe ".aln"
CLUSTAL W (1.8) multiple sequence alignment |
Format GFF / GTF - Ensembl Le format GFF ("General Format Feature") est composé d'une ligne par fonction. Chaque ligne contient 9 colonnes de données et des lignes en optionnelles de définition de piste. Le format GTF ("General Transfer Format") est identique au format GFF version 2. Les champs doivent être séparés par des tabulations. Tous les champs (sauf le dernier) de chaque ligne doivent contenir une valeur : seqname : nom du chromosome (avec ou sans le préfixe « chr ». Ce nom doit être un nom de chromosome standard ou un identifiant Ensembl X Ensembl Repeat 2419108 2419128 42 . . hid=trf; hstart=1; hend=21 X Ensembl Repeat 2419108 2419410 2502 - . hid=AluSx; hstart=1; hend=303 X Ensembl Repeat 2419108 2419128 0 . . hid=dust; hstart=2419108; hend=2419128 X Ensembl Pred.trans. 2416676 2418760 450.19 - 2 genscan=GENSCAN00000019335 X Ensembl Variation 2413425 2413425 . + . X Ensembl Variation 2413805 2413805 . + . |
Format GCC/MSF ("Pileup") - ("Genetics Computer Group" - GCG fondu dans "Accelrys")
1YYCA |
Format PIR/NBRF
1YYCA 174 bases GHHHHHHLEA SADEKVVEEK ASVISSLLDK AKGFFAEKLA NIPTPEATVD DVDFKGVTRD GVDYHAKVSV KNPYSQSIPI CQISYILKSA TRTIASGTIP DPGSLVGSGT TVLDVPVKVA YSIAVSLMKD MCTDWDIDYQ LDIGLTFDIP VVGDITIPVS TQGEIKLPSL RDFF* Ci-dessous : extrait d'un fichier de fonction au format PIR / NBRF utilisé dans la base de données PIR : F;1/Modified site: Removed, cleaved_initiator_methionine |
Format Stanford / IG
;1YYCA 174 bases |
![]()