| Le stockage de la bioinformation : les bases de données |
| Tweet |
|
|
Les fichiers contenant l'information biologique sous la forme de séquences constituent l'élément central autour duquel les bases de données se sont constituées à l'origine. On peut distinguer :
Il existe un très grand nombre de bases de données d'intérêt biologique. Le panorama de ces milliers de bases de données biologiques nécessitent cependant un préalable qui s'appuie sur une forme de "sagesse" :
|
|
1. Les bases de données généralistes Les bases de données généralistes sont indispensables à la communauté scientifique car elles regroupent des données et des résultats essentiels dont certains ne sont plus reproduits dans la littérature scientifique. Exemples de bases de données considérées comme des recueils de référence mondiaux :
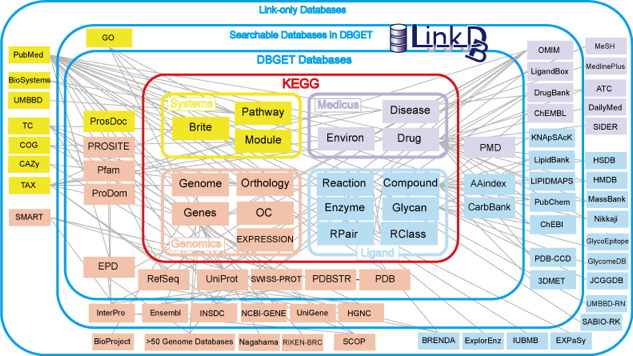
Dans le cadre de l'analyse des séquences, par exemple, le fait que la majorité des séquences connues soit réunie en un seul ensemble est un élément fondamental pour la recherche de similitudes avec une nouvelle séquence. D'autre part, la grande diversité d'organismes qui y est représentée permet d'aborder des analyses de type évolutif. La principale mission des bases de données généralistes est de rendre publiques les séquences et tout autre type d'information. Cette notion de mise à la disposition du public a été capitale dans le cas par exemple de la diffusion des résultats du séquençage du génome humain. On y trouve également de l'information qui accompagne les séquences (annotations, bibliographie, ...) et une expertise biologique directement liées aux séquences traitées. La présence de références à d'autres bases permet d'avoir accès à d'autres informations. Les multiples liens entre les groupes de données dans les bases de données généralistes sont d'une complexité étonnante. Voir les exemples du NCBI. La qualité des données contenues dans ces bases présente un certain nombre de lacunes. Les organismes responsables de la maintenance de ces banques ont pris conscience de la nécessité de vérifications des données soumises ou saisies (surtout pour les séquences anciennes). Maintenant, de nombreuses vérifications sont faites systématiquement dès la soumission de la séquence : c'est la curation dont dépend la fiabilité de l'annotation. Exemple : équipe de C. O'Donovan à l'EBI. Il existe désormais un recueil de séquences référencées, annotées et contrôlées : The Reference Sequence (RefSeq) collection. Des liens sont établis entre les bases de données : certaines d'entre elles échangent même leur données quotidiennement pour actualiser leurs informations. Un autre type de lien est établi via les numéros d'accession.
La figure originale permet de visualiser ces liens et surtout de récupérer, pour un fichier donné, l'ensemble des fichiers équivalents dans les autres bases de données. |
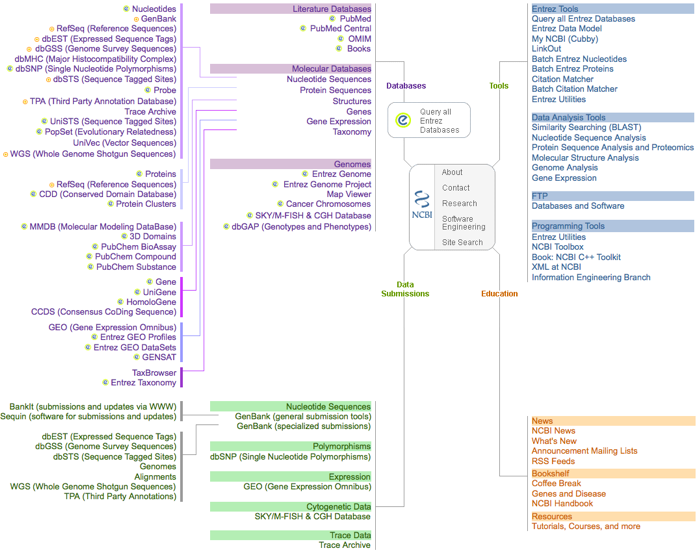
2. Exemple d'une base de données généraliste Genbank - NCBI : Créée en 1982 par la société IntelliGenetics et diffusée maintenant par le NCBI ("National Center for Biotechnology Information", Bethesda - Maryland). Figure ci-dessous : "site map" de l'ensemble de la base de données du NCBI.
Cette figure décrit les relations entre les différentes tables de données et les programmes constitutifs de cet ensemble de plus en plus gigantesque. |
| Type de données biologiques | Base de données | Nombre |
| Séquences protéiques annotées manuellement Nombre d'acides aminés |
UniProtKB/TrEMBL (2021) | ≈ 564.000 ≈ 200 millions |
| Séquences protéiques annotées par programmes Nombre d'acides aminés |
≈ 146 millions ≈ 71 milliards |
|
| Plus de 95% des séquences de protéines de UniProtKB sont issues de la traduction in silico de séquences codantes soumises aux bases de données de séquences nucléotidiques (EMBL-Bank / GenBank / DDBJ), c'est-à-dire à la collaboration des bases de données internationales de séquences nucléotidiques ("International Nucleotide Sequence Database Collaboration" - INSDC). | ||
| Séquences nucléotidiques Nombre de nucléotides |
GenBank (2021) | ≈ 226 millions ≈ 941 milliards |
| Séquences de référence | RefSeq (2021) | ≈ 191 millions |
| Structure 3D (PDB) | PDB ("Protein Data Bank") (2021) | ≈ 182.000 |
| Nombre de repliements ("protein folds") | SCOP ("Structural Classification of Proteins") (2020) |
≈ 1496 |
| Familles de protéines | Pfam (2020) | ≈ 18.300 |
Autres exemples de bases de données généralistes DDBJ ("DNA Data Bank of Japan") : Créée en 1986 et diffusée par le NIG ("National Institute of Genetics", Japon).
Ces grandes bases de données généralistes s'échangent systématiquement leur contenu depuis 1987 et adoptent un système de conventions communes (The DDBJ/EMBL/GenBank Feature Table Definition). PIR-NBRF ("Protein Information Ressource") : banque de protéines créée sous l'influence du NBRF ("National Biomedical Research Foundation") à Washington. Elle diffuse maintenant des données issues du MIPS ("Martinsried Institute for Protein Sequences"), de la base Japonnaise JIPID ("Japan International Protein Information Database") et des données propres de la NBRF. GOLD ("Genomes OnLine Database") : base de données qui recensse les milliers de génomes séquencés ou en voie de séquençage. "Nucleic Acids Research" (NAR) est un exemple de journal scientifique dédié plus particulièrement à la diffusion des bases de données biologiques. |
3. Exemple plus détaillée : ExPASy - Uniprot - PIR/NBRF ExPASy ("Expert Protein Analysis System") est une émanation du SIB ("Swiss Institute of Bioinformatics"). ExPASy permet l'accès à une multitude de bases de données (issues de différents domaines des sciences de la vie - génomique, transcriptomique, protéomique, métabolomique, biologie des systèmes, ...) et d'outils logiciels pour manipuler ces données. ExPASy développe (entre autres) :
|
| Base de données | Nom du service Web / URL vers la documentation |
| NCBI | E-utility Web Service [SOAP ("Simple Object Access Protocol") & WSDL ("Web Services Description Language")] |
| Script Python de recherche bibliographique dans la base de données Pubmed - NCBI (via Entrez et les outils "Eutils"). | |
| EMBL/EBI | EMBL-EBI Web Services - REST / http://www.ebi.ac.uk/Tools/webservices/ |
| UniprotKB | Programmatic access services - REST |
| PDB | Web Services Overview |
| KEGG | REST- style KEGG API |
| REST ("Representational State Transfer") : style d'architecture logicielle; API ("Application Programming Interface") : interface de programmation d'applications. | |
|
4. Les bases de données spécialisées Pour des besoins spécifiques liés à l'activité d'un groupe de personnes, ou encore par compilations bibliographiques, de nombreuses bases de données spécifiques ont été créées au sein des laboratoires. Certaines sont inconnues ou mal connues et attendent qu'on les exploite davantage. Les bases de données spécialisées sont d'intérêt divers et la masse des données qu'elles contiennent peut varier d'une base à une autre. Ces bases correspondent à des améliorations ou à des regroupements par rapport aux données issues des bases généralistes. Exemples de bases de données spécialisées Late Embryogenesis Abundant Proteins database (LEAPdb - Hunault & Jaspard, 2010) : cette base de données contient des informations sur les protéines LEA impliqués dans la tolérance à de nombreux stress, notament la déshydratation et le froid. small Heat Shock Proteins database (sHSPdb - Jaspard & Hunault, 2016) : cette base de données contient des informations sur les protéines de choc thermique de faible masse molaire. RESID Database of Protein Modifications : base de données sur les acides aminés peu fréquents (sous-partie de la base de données PIR). |
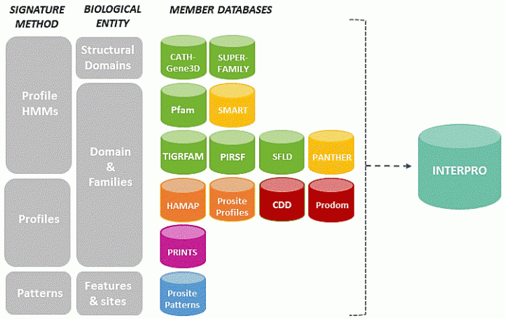
5. Le consortium de bases de données InterPro InterPro permet l'analyse de séquences de protéines en les classant dans des familles et en prédisant la présence de domaines et de sites fonctionnels. InterPro est un consortium : pour mieux classer les protéines, InterPro utilise en effet les modèles ("patterns"), les profils ("profiles") et les signatures ("fingerprints") fournis par 14 bases de données membres (regroupées en une seule ressource) : CATH-Gene3D, SUPERFAMILY, Pfam, SMART, TIGRFAM, PIRSF, SFLD, PANTHER, HAMAP, Prosite, CDD, MobiDB, ProDom, PRINTS.
Source : InterPro Par exemple, SUPERFAMILY est une base de données d'annotations structuralles et fonctionnelles des protéines et des génomes. C'est une bibliothèque de modèles de Markov cachés de profils : chaque modèle représente un domaine protéique de structure connue au niveau d'une superfamille SCOP (qui regroupe des domaines qui ont une relation évolutive). L'annotation est générée en confrontant les séquences protéiques de plus de 3200 génomes complètement séquencés contre ces modèles de Markov cachés. Exemple de classification dans la base de données InterPro (les liens renvoient vers chaque niveau de classification mentionnée) :
|
6. La base de données Pfam (&asymp 18.300 familles - 2020) La possibilité de diverses combinaisons de multiples domaines explique la très grande multiplicité des protéines. La caractérisation du ou des domaines d'une protéine permet d'en décrypter la/les fonction(s).
Source : Prosite La base de données Pfam est une collection de familles de domaines des protéines : chaque famille est représentée par des alignements multiples des séquences et un modèle de Markov caché ("hidden Markov model" - HMM). Chaque famille ou entrée Pfam (souvent désignée sous le nom "Pfam-A entry") est constituée d'un alignement de séquences généré de la manière suivante :
Script python de recherche du profil HMM d'une famille PFAM. Les entrées Pfam sont classées en 6 catégories, en fonction de la longueur et de la nature des parties de la séquence inclues dans l'entrée :
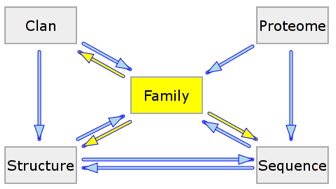
Plusieurs entrées Pfam liées sont regroupées dans un clan. Leur inter-relation est définie par :
Source : Pfam |
7. Les bases de données de motifs L'utilisation de bases spécialisées comme les bases de motifs est devenue un outil essentiel dans l'analyse des séquences pour tenter de déterminer la fonction de protéines inconnues ou savoir à quelle famille appartient une séquence non encore caractérisée. a. Les bases de motifs nucléiques La plupart de ces bases consiste à recenser dans des catalogues les séquences des différents motifs pour lesquels une activité biologique a été identifiée. Certains motifs sont simples et non ambigus, d'autres correspondent à des activités biologiques plus complexes et engendrent donc des séquences moins précises. Pour ces derniers types de motifs, des compilations ont été établies pour donner des listes annotées de motifs qui peuvent être communs à plusieurs séquences. Il existe différentes bases de motifs nucléiques, notamment celles concernant les motifs de fixation des facteurs de transcription. b. Les bases spécialisées de motifs protéiques La base PROSITE peut être considérée comme un dictionnaire qui recense des motifs protéiques ayant une signification biologique. Elle est établie en regroupant, quand cela est possible, les protéines contenues dans Swissprot par famille (Exemples : les kinases ou les protéases). On recherche ensuite, au sein de ces groupes, des motifs consensus susceptibles de les caractériser spécifiquement. La conception de la base PROSITE repose sur quatre critères essentiels :
Voir un exemple : motif "EF-hand" des protéines fixant le calcium comme la calmoduline par exemple. |
c. Exemples de logiciels et bases de données de profils PSSM Voir un cours sur les profils et matrices PSSM ("Position Specific Scoring Matrice"). Pftools : ensemble d'outils logiciels (« package ») pour construire des profils dans le but de rechercher des séquences et les aligner. Parmi ces programmes :
PRINTS : base de données de profils PSSM.
PRINTS est l'un des partenaires fondateurs du consortium de ressources bioinformatiques InterPro (base de données de familles de protéines, de domaines et de sites fonctionnels).
Script Python pour la recherche du profil HMM d'une famille PFAM. |
| 8. Liens Internet et références bibliographiques |
| Base de données sur les acides aminés peu fréquents (sous-partie de la base de données "Protein Information Resource" - PIR) | |
| Bases de données sur les propriétés physico-chimiques des acides aminés (sous-partie de la base de données "Expasy - Swiss-Prot") | |
| NAR Database Summary Paper Category List | |
|
Hunault G. & Jaspard E. (2010) "LEAPdb: a database for the late embryogenesis abundant proteins" BMC Genomics 11, 221 Jaspard E. & Hunault G. (2016) "sHSPdb: a database for the analysis of small Heat Shock Proteins" BMC Plant Biol. 16, 135 |
![]()