
| Génomique : les méthodes de séquençage d'acides nucléiques et l'acquisition des données |
| Tweet |
|
|
1. Introduction a. Deux types de molécules support de la bioinformation : les acides nucléiques et les protéines 2. Détermination des séquences de nucléotides a. Méthode historique de Fréderick Sanger 3. Méthode du pyroséquençage 4. Stratégies initiales de séquençage des génomes : méthode hiérarchique vs. méthode en "vrac" 5. Les contigs et l'assemblage, les trous, l'appel de base 6. Les nouvelles technologies de séquençage à trés haut débit ("next-generation high-throughput DNA sequencing technologies" - NGST ou NGS)
|
b. Caractéristiques élémentaires des NGS 7. Quelques exemples d'apports du séquençage 8. Etude des éléments de la régulation de la transcription - Structure de la chromatine et épigénomique 9. Epigénétique - modifications de l'ADN et des histones 10. Séquençage du transcriptome - RNAseq a. Méthodes pour l'assemblage des lectures 11. Liens Internet et références bibliographiques |
|
1. Introduction a. Deux types de molécules support de la bioinformation : les acides nucléiques et les protéines Le "matériaux de base" de la génomique et de la protéomique est la séquence : l'enchaînement ordonné et orienté de nucléotides (acides nucléiques) ou d'acides aminés (protéines). |
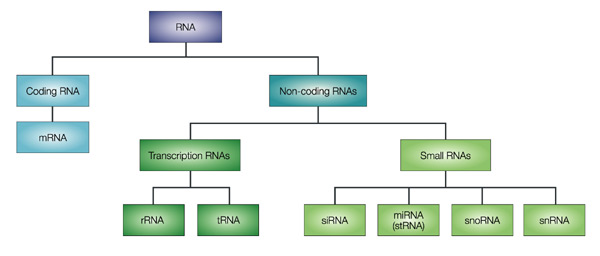
ADN : Acide DésoxyriboNucléique
On distingue :
|
|
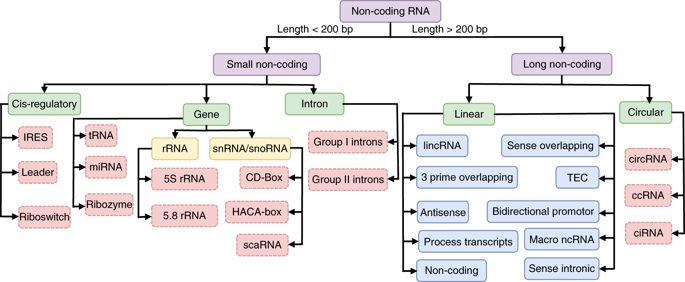
ARN : Acide RiboNucléiques
On distingue :
|
Protéines
|
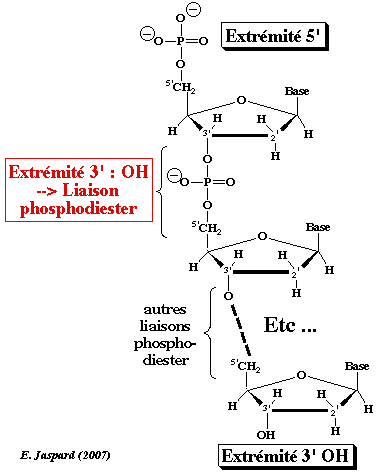
Les chaînes nucléotidiques possèdent 2 extrémités distinctes : on peut donc les représenter de manière orientées de l'extrémité dite 5' vers l'extrémité dite 3'. En conséquences, les chaînes nucléotidiques sont écrites sous forme d'une succession ordonnée et orientée de lettres qui représentent les unités élémentaires (les nucléotides) :
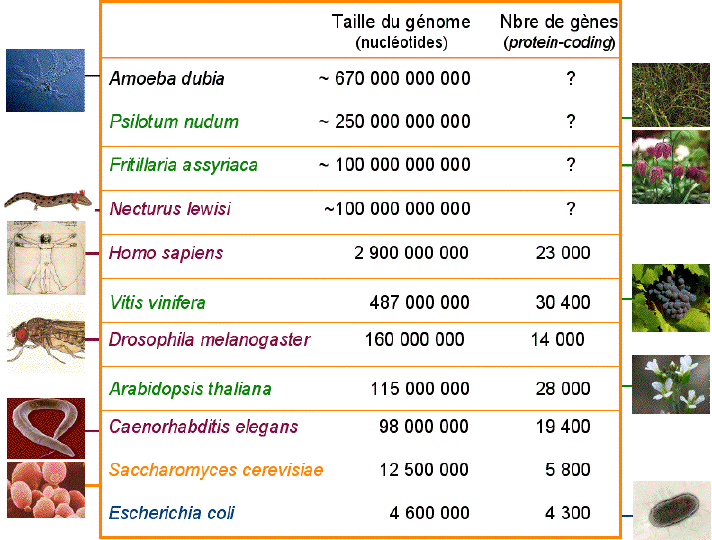
La taille des génomes nucléaires varie énormément au sein des Eucaryotes :
L'un des plus célèbres génome séquencé est celui de l'homme de Neandertal (Green et al., 2010).
Source : B. Dujon (2008) GOLD ("Genomes OnLine Database") : base de données des génomes séquencés et en cours de séquençage. |
|
b. "Préhistoire" du séquençage des acides nucléiques et séquençage dans l'espace Un énorme effort humain, financier, technologique, a été fait dans les années 90 pour obtenir des outils pour les premiers pas du séquençage, de plus en plus performants et surtout automatisés. Pour le séquençage des premiers génomes "historiques" (entre autre le génome humain), l'automatisation a requis dans les années 1990 / 2000 le développement :
Source : Nature 409, 860 - 921 Ci-dessous : en 2016, un séquenceur Illumina.
Capacité de séquençage : 5 milliards de lectures x [300 paires de bases] = 1500 miliards de nucléotides en quelques heures à 1 jour. Ci-dessous : le séquenceur ultra-portable MinION (Oxford nanopore technology) - dit de 3è génération - a été utilisé en temps réel sur le terrain lors de la crise Ebola de 2015 et de la crise Zika en 2016 (Quick et al., 2016).
Source : The guardian Les dernières avancées Les technologies avec des nanopores sont de plus en plus performantes. En 2018, le séquençage et l'assemblage de novo d'un génome humain s'est appuyé sur un protocole :
En juillet 2016, la technologie Minion a été envoyée par la NASA dans la station spatiale internationale ("International Space Station", ISS) pour les premiers séquençages effectués dans l'espace d'ADN extra-terrestre potentiel. Elle a été testée avec succès dans des conditions de gravité comparables à celles qui règnent sur Mars (G = 0,378), sur la lune (G = 0,166) et sur Europa (sattelite de Jupiter - G = 0,134). Voir : Carr et al. (2020). Le séquençage en routine du génome humain est devenu possible avec le séquenceur PromethION (Oxford Nanopore Technologies) qui possède 3000 capteurs et 12.000 pores : ils génèrent en moyenne 70 Go de données permettant une couverture 20 X du génome humain. Enfin, la précision des logiciel d'appel de base est sensiblement améliorée par des algorithmes basés sur des modèles de Markov cachés ou des réseaux de neurones. |
|
1953: séquençage de l'insuline (Frederick Sanger) 1965: séquençage de l'ARNt alanine 1968: séquençage des extrémités cohésives de l'ADN du phage lambda 1977: technique de séquençage de l'ADN de Allan Maxam & Walter Gilbert 1977: technique de séquençage de l'ADN de Frederick Sanger 1981: vecteur du phage M13 de Messing 1986: Détection des bases par fluorescence au cours du séquençage par électrophorèse 1987: sequenase 1988: premier séquençage par incorporation progressive de dNTP 1990: séquençage par extrémités appariées 1992: colorants Bodipy 1993: colonies d'ARN in vitro 1996: pyroséquençage 1999: colonies d'ADN dans des gels 2000: séquençage massivement parallèles de signatures par ligation |
2003: PCR en émulsion pour générer des colonies d'ADN sur des billes |
| Source : Shendure et al. (2017) | |
2. Détermination des séquences de nucléotides a. Méthode historique de Fréderick Sanger Fréderick Sanger est décédé en 2013. Il fût l'un des plus admirables scientifiques - biochimistes (Prix Nobel de chimie 1958 et Prix Nobel de chimie 1980). |
| Bien qu'elle ait cédé la place aux nouvelles technologies de séquençage, la méthode de Sanger est historiquement capitale puisqu'elle a permis les premiers séquençages de génomes complets : | |
|
|
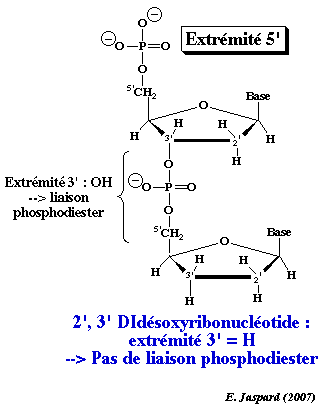
Les nucléotides au sein des acides nucléiques sont liés par une liaison phosphodiester qui s'établit entre le groupement OH sur le carbone 3' du ribose du nucléotide dit en position 5' et le phosphore du groupe phosphoryle en position α du nucléotide dit en position 3'. La méthode de séquençage de Sanger (dite par terminaison de chaîne) utilise des nucléotides appelés didésoxyribonucléotides (ddNTP) qui ont un atome d'hydrogène à la place du groupement OH sur le carbone 3' du ribose. Ils peuvent donc être incorporés dans un brin d'ADN en cours de synthèse, mais ils ne permettent pas qu'un autre nucléotide soit incorporé après eux : en effet, l'absence de l'atome d'oxygène en 3' empêche la formation d'une nouvelle liaison phosphodiester. L'allongement du brin d'ADN s'arrète donc au niveau du ddNTP incorporé, d'où terminaison de la synthèse de l'ADN. |
|
|
Sanger et al. (1977) Proc. Natl Acad. Sci. 74, 5463-5467 |
La méthode de séquençage de Sanger utilise une amorce marquée radioactivement ("dye-labeled primer") car la polymérase nécessite un court fragment complémentaire du brin à séquencer pour initier la synthèse du brin copie. Quatre réactions de séquençage sont donc menées en parallèle dans quatre tubes distincts, contenant chacun un seul didésoxyribonucléotide (ddTTP, ddATP, ddCTP et ddGTP) :
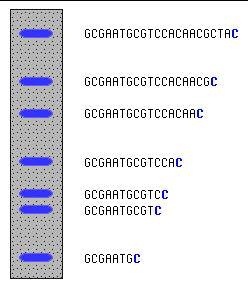
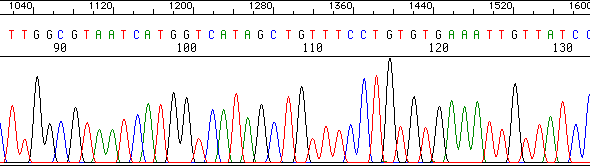
Dans chaque tube, toutes les copies d'ADN synthétisé sont interrompues derrière le même nucléotide. Le rapport des concentrations entre les dNTP et les didésoxyribonucléotides (ddNTP) et le nombre de réactions simultanées catalysées par la polymérase assure statistiquement que toutes les copies partielles intermédiaires possibles de la molécule d'ADN sont synthétisées. On sépare alors les copies selon leur taille par une migration électrophorétique dans un gel poreux (entre 2 larges plaques de verre), le contenu de chaque tube étant déposé dans un puits distinct. Ces gels permettent de séparer deux intermédiaires consécutifs qui ont une différence de taille d'un seul nucléotide. Exemple ci-dessous : profil d'électrophorèse du contenu du tube avec le ddCTP. Toutes les copies intermédiaires d'ADN synthétisé sont terminées par un C.
Source : University of Michigan |
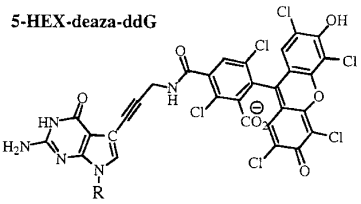
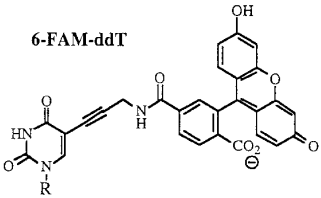
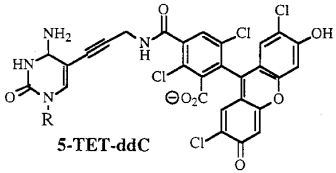
b. La technique de séquençage avec des didésoxyribonucléotides fluorescents ("dye terminator sequencing") Smith et al. (1986) "Fluorescence detection in automated DNA sequencing" Nature 321, 674 - 679 Cette technique utilise des didésoxyribonucléotides dont chacun est marqué par un fluorophore spécifique. Les fragments d'ADN synthétisés portent ce fluorophore terminal. On les appelle des terminateurs d'élongation ou "Big Dye Terminators" ou "Dye-labeled terminator". Ci-dessous, exemple de structures de ddNTP fluorescents :
Source : Brandis (1999) Nucleic Acids Res. 27, 1912-1918
Améliorations apportées par la méthode des ddNTP fluorescents par rapport à la méthode de Sanger a. La méthode initiale de Sanger utilisant une amorce marquée radioactivement est plus laborieuse, coûteuse (4 réactions distinctes) et dangereuse (radioactivité) que celle des ddNTP fluorescents. b. Par ailleurs, l'un des problème du séquençage est la formation de "faux-stop" : c'est la terminaison prématurée d'une copie qui implique un désoxyribonucléotide à la place d'un ddNTP. Avec la méthode des ddNTP fluorescents, les "faux-stop" ne sont pas détectés car ils ne fluorescent pas. c. Avec la méthode des ddNTP fluorescents, il n'y a qu'une réaction de séquençage en présence des 4 didésoxyribonucléotides : ADN matrice + dNTP + ddCTP fluorescent bleu + ddATP fluorescent vert + ddGTP fluorescent jaune + ddTTP fluorescent rouge
Source : University of Michigan |
Ci-dessous, le séquenceur "MegaBACE" (société Amersham) : plateforme capillaire à haut débit pour le séquençage d'ADN.
Schématiquement, l'appareil est composé de 96 capillaires, d'un système d'électrophorèse, d'un laser et d'une caméra CCD (Charge-Coupled Device).
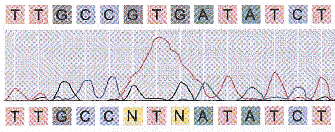
La dernière étape est la lecture des profils bruts ou "base-calling" (détermination de la séquence par appel de bases).
|
|
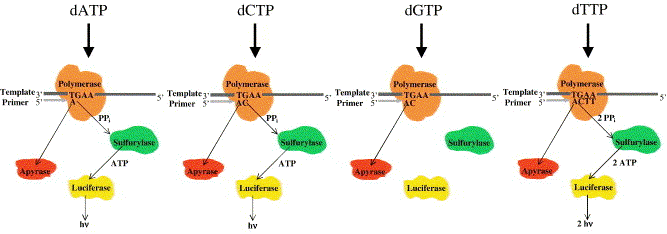
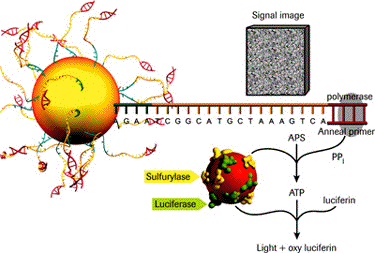
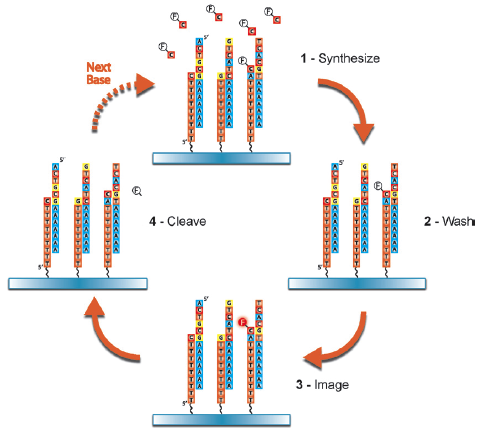
Elle permet d'effectuer un séquençage moins cher et rapide qu'un séquençage par la méthode de Sanger car elle ne nécessite pas de clonage et la lecture de la séquence est directe.
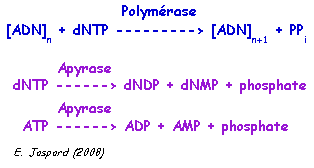
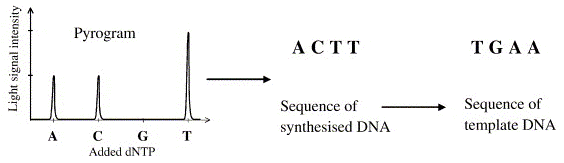
Source : Ahmadian et al. (2006) Les désoxyribonucléotides triphosphate (dNTP) sont ajoutés l'un après l'autre (et non pas tous ensemble comme dans la méthode de Sanger). Si le désoxyribonucléotide ajouté est complémentaire du désoxyribonucléotide du brin matrice, il est incorporé dans le brin en cours de synthèse et un pyrophosphate inorganique (PPi) est libéré.
Le capteur CCD du séquenceur capte le signal lumineux et le traduit par un pic sur le pyrogramme.
Source : Ahmadian et al. (2006) La hauteur du pic est proportionnelle à l'intensité du signal lumineux, elle-même proportionnelle au nombre de nucléotides incorporés au même moment. On déduit la séquence à partir de la taille des pics obtenus. En cas de mélange de nucléotides à une même position (polymorphisme de séquence), la taille des pics permet d'avoir une quantification de la proportion de brins porteurs de l'un ou l'autre des nucléotides. Exemple d'application du pyroséquençage à l'étude du transcriptome de Arabidopsis thaliana : Weber et al. (2007) "Sampling the Arabidopsis transcriptome with massively parallel pyrosequencing" Plant Physiol. 144, 32-42. |
| Méthode | longueur des lecture (nucléotides) | nombre de lectures | total par tour ("run") (Mpb) | coût relatif par nucléotide |
| Sanger | 700 - 800 | 96 | 0,07 | 1 |
| pyroséquençage | 250 | 400.000 | 100 | 0,1 |
| phase solide | 25 - 35 | 40 à 80 millions | 1000 - 2000 | 0,01 |
Cette partie retrace un "historique" des stratégies de séquençage avant l'avènement des nouvelles technologies de séquençage à très haut débit. En regard de l'avancée phénoménale des techniques et des capacités d'analyse, certaines parties peuvent sembler obsolètes mais elles représentent certains fondements de la génomique. |
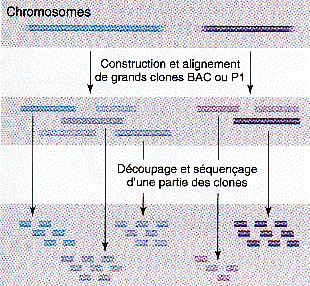
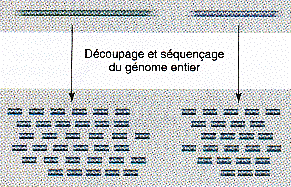
b. La méthode de séquençage aléatoire global ou "en vrac" ou "shotgun" C'est une méthode très différente et complémentaire de la méthode hiérarchique. Une carte de grands fragments ordonnés n'est pas établie au préalable. Un trés grand nombre de séquences sont obtenues de façon aléatoire à l'échelle du génome entier. Les extrémités d'une partie de ces fragments sont séquencées. Puis ces séquences sont assemblées selon leurs recouvrements. Du fait du grand nombre de fragments et du clonage, certaines séquences ne sont jamais séquencées.
Source : "Précis de génomique" Gibson & Muse (2004) La difficulté d'assemblage est beaucoup plus grande que dans la stratégie "clone par clone" et le nombre énorme de comparaisons de séquences nécessite une puissance de calcul considérable. Il n'est pas possible, pour combler les trous entre les contigs (voir ci-dessous), de diriger le travail de séquençage supplémentaire sur un grand fragment bien identifié. Compléments sur la méthode "shotgun" C'est un processus aléatoire d'échantillonnage de N lectures de taille L, pour un génome de taille G :
Lander & Waterman (1988) "Genomic mapping by fingerprinting random clones: A mathematical analysis" Genomics 2 , 231 - 239 Evolution des "stratégies" de séquençage de type "shotgun" :
|
Quelle que soit la stratégie adoptée, lors de l'assemblage terminal du génome, il faut éliminer :
|
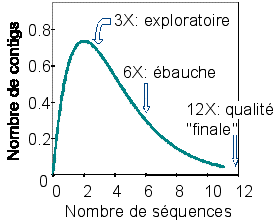
5. Les contigs et l'assemblage, les trous, l'appel de base Avec les technologies encore courantes dans de nombreux laboratoires, chaque séquençage ne permet d'obtenir une lecture que de quelques milliers de paires de base. Il n'est donc pas possible de séquencer en une seule fois des molécules d'ADN aussi grandes que les chromosomes. Pour reconstituer ces immenses séquences, il faut effectuer un grand nombre de séquençages, plusieurs fois supérieur à la taille du chromosome. Ces séquençages redondants permettent :
Pour les premiers séquençages des génomes (avant l'avènement des nouvelles technologies de séquençage à très haut débit), la redondance était d'un facteur 8 à 10 (une profondeur de 8 à 10X).
Source : B. Dujon (2008) Celà signifie :
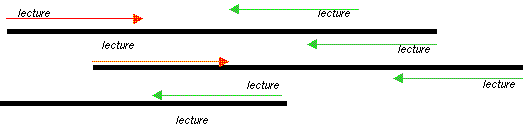
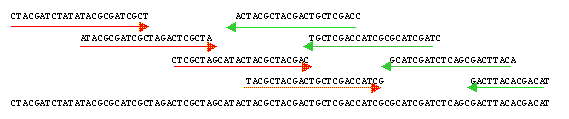
L'assemblage La comparaison des séquences permet d'aligner les parties qui se recouvrent partiellement ou chevauchantes.
Source : Genoscope - FAQ Les séquences chevauchantes peuvent être reliées en enchaînements plus grands que l'on appelle contigs. En reliant l'ensemble des contigs, on reconstitue des séquences de plusieurs millions à plusieurs dizaines de millions de nucléotides (les "scaffold"). Ces opérations sont effectuées par des programmes bioinformatiques. Les trous ou "gap" : Comme le séquençage est effectué sur des sous-fragments pris de manière aléatoire, même avec un tel niveau de redondance, il reste des parties non assemblées : des trous ("gap") qui peuvent être "comblés" par un travail ciblé. Scaffold : ensemble de contigs orientés et ordonnés. Les trous ("gaps" - voir ci-dessous) sont de longueur connue. Mapped scaffold : ensemble de scaffolds localisés le long des chromosomes (pas forcément ordonnés ou orientés). Les trous sont de longueur inconnue. Pour déterminer les relations de voisinage des contigs, les liens clones sont considérés, c'est-à-dire les lectures obtenues aux deux extrémités d'un même fragment d'ADN. On recherche parmi ces paires celles qui s'ancrent dans deux contigs différents. Cela permet de jeter un pont entre les deux contigs et de les orienter. De plus, le fragment d'ADN "à cheval" sur le trou entre les deux contigs peut faire l'objet d'un séquençage supplémentaire, ce qui permet de combler le trou. La lecture des profils bruts ou "base-calling" : c'est la détermination de la séquence par appel de bases qui s'effectue en routine par des programmes informatiques qui déterminent l'identité des bases, comparent les séquences et fournissent une plate-forme intuitive de correction. La suite logicielle publique développée à l'Université de Washington contient les programmes :
Source : "Précis de génomique" Gibson & Muse (2004) |
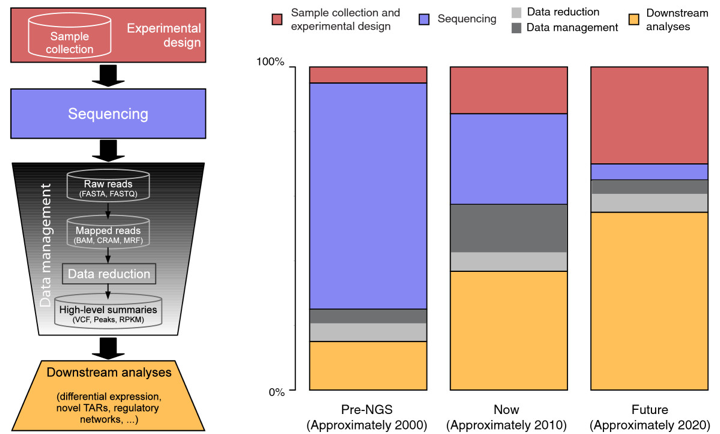
6. Les nouvelles technologies de séquençage à trés haut débit ("next-generation high-throughput DNA sequencing technologies" - NGST ou NGS) Une révolution en génomique fonctionnelle a eu lieu avec l'avènement des technologies de séquençage à trés haut débit ou massivement parallèles. Des quelques 800 à 1000 nucléotides qu'un chercheur pouvait espérer séquencer en quelques jours par des techniques lourdes, complexes et dangereuses (utilisation d'isotopes radioactifs) dans les années 80, on est arrivé à l'heure actuelle à des techniques de séquençage simplifiées qui séquencent des milliards de nucléotides par expérience. L'ensemble des données de séquençage est implémenté en temps réel dans des bases de données pour leur analyse. En conséquence, de plus en plus de génomes sont séquencés ou en cours de séquençage avec l'avènement des nouvelles technologies de séquençage à trés haut débit. Janvier 2020
Projets de très grande envergure de séquençage de génomes
Des biologistes et des informaticiens prévoient que les ressources informatiques nécessaires pour traiter les données liées aux génomes dépasseront à terme celles nécessaires à Twitter et YouTube. On estime que, en 2025, 100 millions à 2 milliards de génomes humains auront été séquencés. A lui seul, le stockage de ces données pourrait nécessiter 2 à 40 exaoctets (1 exaoctet = 1018 octets) car les données stockées pour un génome sont 30 fois plus grande que la taille du génome lui-même (données brutes, erreurs, analyse préliminaire …). Le stockage des données ne sera qu'une petite partie du problème : les besoins pour l'acquisition, la distribution et l'analyse des données de génomiques seront bien supérieurs. |
|
b. Caractéristiques élémentaires des NGS
Le séquençage de novo : c'est le séquençage d'un génome pour la première fois. Il nécessite l'assemblage d'un très grand nombre de petites séquences du génome. Il nécessite aussi un génome de référence (s'il en existe un) afin de positionner (par comparaison) les séquences obtenues. Définitions importantes
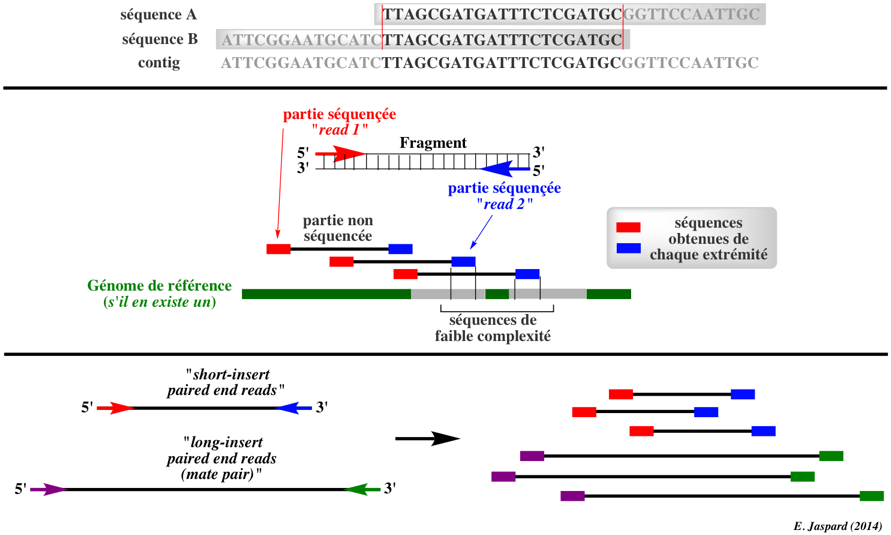
Le séquençage complet d'un génome avec les NGS conduit à un nombre colossal de petits fragments séquencés (un grand nombre de petites séquences ou lectures) que l'on essaye ensuite d'assembler en contigs. La qualité de couverture du séquençage et donc liée à celle des contigs (leur longueur et leur continuité) et donc au nombre de gaps. L'un des inconvénients des NGS est la petite taille des fragments séquencés d'où un nombre élevé de gaps, en particulier pour les régions de faible complexité.
a. Pour pallier à cette difficulté, on peut séquencer les fragments :
b. Un autre moyen est de construire des banques avec des inserts de petites tailles (0,2 - 0,8 kpb) et des banques avec des inserts de grandes tailles (2 - 40 kpb). On obtient ainsi des fragments séquencés de tailles variables ("short-insert paired end reads" et "long-insert paired end reads" ou "mate paired") qui aboutissent à un meilleur assemblage du fait de contigs plus longs. L'acquisition et la compilation d'une masse de données de plus en plus astronomique d'une part et l'analyse des résultats des NGS nécessitent le développement d'outils bioinformatiques de plus en plus spécialisés (exemple : Allpaths-LG propose un assembleur pour grands génomes). |
Les NGS permettent d'aborder (liste non-exhaustive) :
Source : Shendure & Aiden (2012)
|
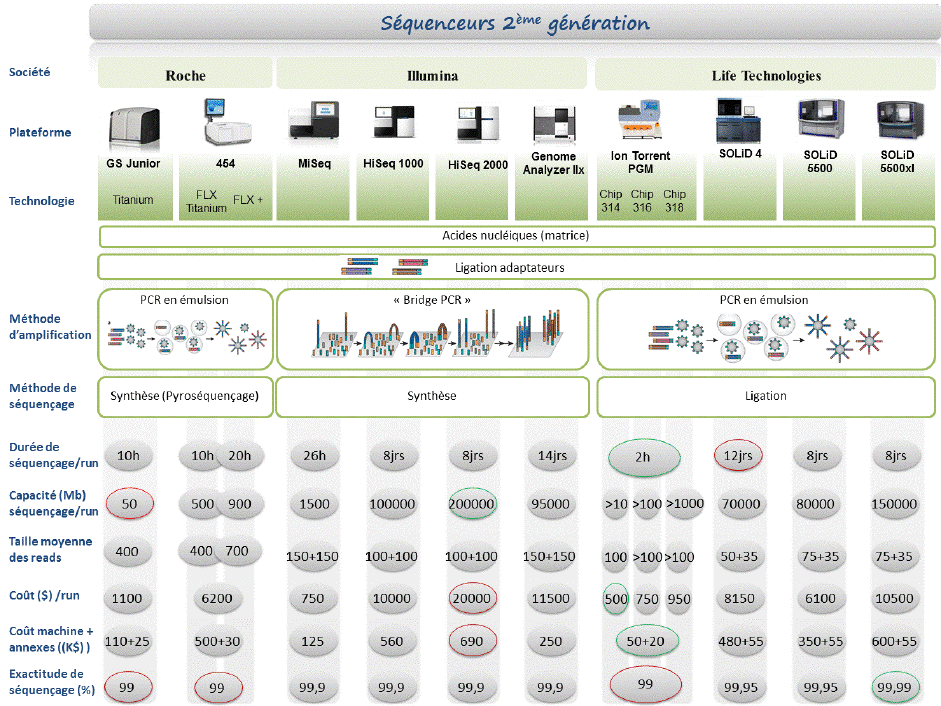
c. Les technologies NGS et les entreprises actuelles De nouvelles technologies apparaissent chaque année (plus puissantes, plus rapides, plus économiques, ...). Il est illusoire de les lister : ne sont donc présentées que celles qui ont permis les premiers grands bonds. 1. "llumina sequencing" : Illumina représente environ 56% du marché des NGS.
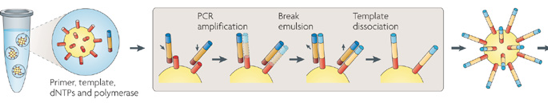
Historiquement, l'entreprise s'est d'abord appelée Solexa (1998) avec un modèle de séquenceur ("Genome Analyzer") lancé en 2006. En 2007, la société Illumina a fait l'acquisition de Solexa. 2. La technologie Roche 454 La société "454 Life Sciences" (Connecticut - USA) a développé les séquenceurs GS20 et GS FLX ("Genome Sequencer" - distribution par Roche Diagnostics). La technique utilisée (Margulies et al., 2005) est basée sur l'amplification d'ADN lié à une bille en émulsion et au pyroséquençage. Voir un film qui décrit le principe de cette méthode de séquençage.
Source : 454.com Exemples d'application
|
| Comparaison des technnologies Illumina et 454 | ||
| Illumina (ex-Solexa) | 454 (Roche) | |
| Amplification en phase solide - séquençage par synthèse (mesure de fluorescence) | Amplification dans une émulsion - pyroséquençage |
|
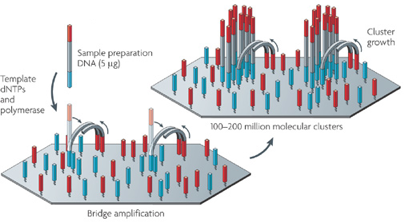
L'ADN génomique est fragmenté. |
L'ADN génomique est fragmenté (300 - 800 paires de bases). |
|
|
Source des figures : Metzker M. (2010) |
|
| Un nucléotide marqué par une étiquette fluorescente ("fluorescently labeled chain-terminating nucleotide") est incorporé dans la chaîne en cours de synthèse (synthèse du brin complémentaire de celui qui est séquencé). La fluorescence de ce nucléotide est mesurée. Mais l'incorporation de ce terminateurs de chaîne est réversible, ce qui permet que la synthèse continue jusqu'à ce qu'un autre terminateur de chaîne soit incorporé. |
Une émulsion (mélange eau-huile) encapsule cette bille et forme une goutelette : l'amplification PCR du fragment d'ADN en milliers de copies s'effectue dans cette goutelette. |
|
| Faible taux d'erreurs d'insertion/délétion (indel). | Taux plus élevé d'insertion/délétion (indel) que la technologie Illumina. |
|
| 1 milliard de bases séquencées (lectures d'environ 30-40 bases - les terminateurs de chaîne réversibles sont incorporés moins efficacement) avec une exactitude moindre que la technologie 454. | 1 million de bases séquencées (lectures plus longues d'environ 250 bases) avec une exactitude de 99.5%. |
|
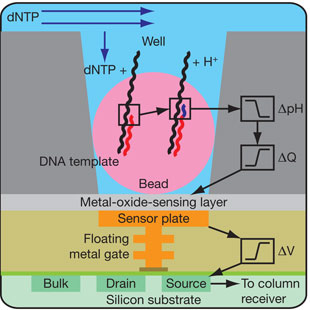
3. La technologie "Ion Torrent" Cette technologies est basée sur des puces semi-conductrices remplies de puits.
Un proton est relargué quand un nucléotide est incorporé par la polymérase dans l'ADN. Cela résulte en un changement de pH local qui est détecté par cette technologie sensible à la variation d'ions.
Voir une vidéo qui décrit cette technologie. Cette technologie ne nécessite pas de camera, pas de scanner, pas de cascade enzymatique, pas de fluorophore ou chemiluminescence.
Source : Rothberg et al. (2011) "An integrated semiconductor device enabling non-optical genome sequencing" Nature 475, 348 - 352 |
4. La technologie SOLiD ("Sequencing by Oligonucleotide Ligation and Detection") : le séquençage est basé sur l'amplification par émulsion et l'hybridation-ligature chimique. Il utilise une ligation avec une DNA ligase. Voir un développement de la technique et du principe des réactions chimiques. 5. La technologie "Helicos BioSciences" Remarque : cette entreprise phare a fait faillite en novembre 2012. Les nucléotides fluorescents sont ajoutés l'un après l'autre. Les nucléotides non incorporés (selon le brin matrice) sont éliminés. Une illumination avec un faisceau laser induit une émission de fluorescence aux endroits où le nucléotide a été incorporé. Le groupe fluorescent du nucléotide qui vien dêtre incorporé est à son tour éliminé afin que le nucléotide suivant puisse être incorporé par la polymérase.
Source : Helicos BioSciences |
d. Les technologies NGS en développement et à venir 1. G4-seq : les quadruplexes G sont des structures secondaires d'acides nucléiques qui se forment au niveau de séquences d'ADN ou d'ARN riches en guanine. Ces structures peuvent affecter l'architecture de la chromatine et la régulation de la transcription des gènes et elles sont associées à l'instabilité des génomes. 2. FIS-seq ("Fluorescence In Situ RNA-sequencing"): le perfectionnement de la technique FISH ("RNA Fluorescence In Situ Hybridization") permet désormais de détecter les molécules d'ARN individuelles dans une seule cellule ("single cell") et d'obtenir des informations sur la localisation sub-cellulaire de ces molécules d'ARN. La technique FIS-seq combine l'approche RNA-seq "classique" (en utilisant la cellule comme une "puce" de séquençage) avec la technique FISH. 3. La spectromètrie de masse appliquée aux fragments d'ADN. 4. La visualisation directe de molécule d'ADN par microscopie de force atomique. Les technologies de séquençage dites de troisième génération Les technologies de séquençage dites de troisième génération permettent d'acquérir de très longues lectures ("ultra-long reads").
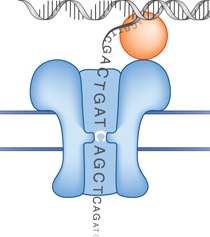
Les longues lectures permettent le chevauchement de longues répétitions et donc l'orientation des contigs avec moins d'ambiguïté, ce qui est essentiel pour l'assemblage de novo de génomes. Illustration : technologie MinION™ (Oxford Nanopore Technologies)
Source : Oxford Nanopore Le séquençage s'effectue via le passage de fragments d'ADN au travers des nanopores constitués de protéines (hémolysine) incluses dans une bicouche lipidique.
Source : Rusk (2013)
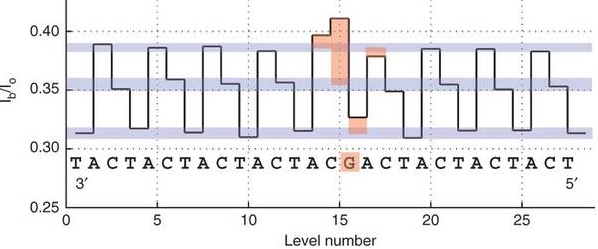
Exemple : l'ADN testé (figure ci-dessous) est constitué de répétitions "CAT" à l'exception d'un triplet "CAG" au milieu de la séquence.
Source : Manrao et al. (2012) La trace du courant moyen montre un profil répété de 3 niveaux (barres bleues) dont la régularité est interrompue au niveau du G (en orange) : les 4 niveaux de courant autour du G sont affectés. Le courant résiduel est donc influencé par 1 ou 2 nucléotides. Les dernières avancées Les technologies avec des nanopores sont de plus en plus performantes. En 2018, le séquençage et l'assemblage de novo d'un génome humain s'est appuyé sur un protocole :
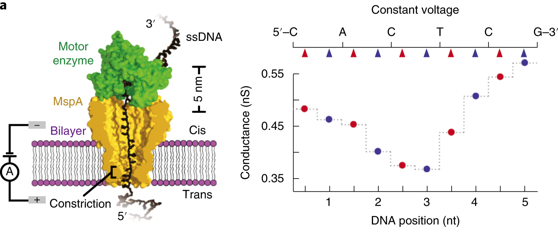
Le séquençage en routine du génome humain est devenu possible avec le séquenceur PromethION (Oxford Nanopore Technologies) qui possède 3000 capteurs et 12.000 pores : ils génèrent en moyenne 70 Go de données permettant une couverture 20 X du génome humain. Enfin, la précision des logiciel d'appel de base est sensiblement améliorée par des algorithmes basés sur des modèles de Markov cachés ou des réseaux de neurones. On envisage d'appliquer la technique des nanopores au séquençage des protéines : la grande difficulté est de déplier la chaîne polypetidique. Une piste est l'utilisation de la "AAA+ unfoldase ClpX". Principe de base du séquençage de nanopores à tension variable Figure de gauche ci-dessous : un nanopore de MspA (Mycobacterium smegmatis porin A) est inséré dans la bicouche lipidique séparant ainsi 2 chambres (cis et trans)/ L'enzyme motrice hélicase Hel308 contrôle le mouvement du brin d'ADN à travers le pore et une tension est appliquée à travers la bicouche : la conductance à travers le pore est mesurée. Figure de droite ci-dessous : séquençage obtenu quand une tension constante est appliquée.
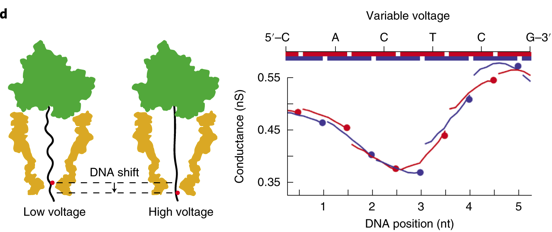
Source : Noakes et al. (2019) Figure de gauche, ci-dessous : l'application de différentes tensions (100 mV à 200 mV) modifie la force de traction appliquée à l'ADN. Des forces élevées entraînent une extension supplémentaire de l'ADN, ce qui a pour conséquence de déplacer l'ADN dans la constriction du pore et ainsi de changer les nucléotides (le point rouge) qui affectent la conductance.
Source : Noakes et al. (2019) Figure de droite, ci-dessus : le séquençage à tension variable analyse la conductance du pore bloqué par l'ADN de manière continue le long de la séquence d'ADN (en haut).
Voir un article de synthèse de l'historique de la technologie ONT, de son évolution, de ses capacités de séquençage (notamment en terme de longueur de "reads" séquencés) et de ses application multiples : Wang et al. (2021) |
|
Le marché du séquençage a été de 1,3 milliard de dollars en 2012. Il est estimé à 2,7 milliards de dollars en 2017. Il pourrait atteindre 28 milliards de dollars en 2022. des caractéristiques des nouvelles technologies de séquençage
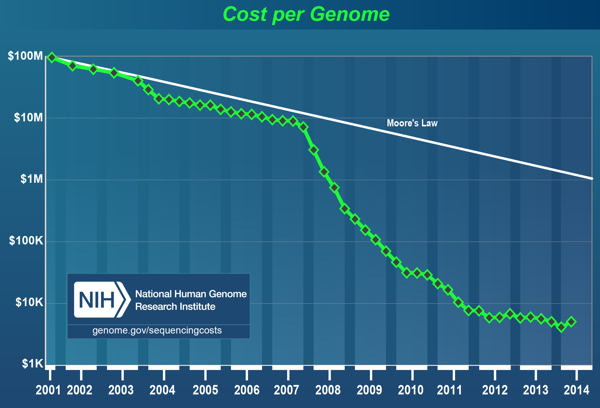
Source : biorigami.com Du fait des données en masse liées aux nouvelles technologies de séquençage à très haut débit, des "simulateurs" ont été développés pour prédire le coût et la durée (estimés sur la base du génome d'Arabidopsis thaliana et de son annotation). Côut du séquençage du génome de l'homme (publié en 2001) : environ 100 millions de dollars. Au cours des premières années, la réduction du côut des séquençages diminuait (diminution ressemblant approximativement à la "Loi - ou énoncé - de Moore").
Janvier 2008 : chute drastique du côut des séquençages avec l'avènement des nouvelles technologies de séquençage à très haut débit, dites de "seconde génération". Depuis le côut baisse régulièrement : il est actuellement d'environ 5000 dollars. On s'approche d'un séquençage complet du génome de l'homme à 1000 dollars. Dans les années à venir, c'est le côut de l'analyse bioinformatique en aval du séquençage d'un génome qui sera prédominante.
Source : Sboner et al. (2011) Acronymes de la figure : AM : Binary Sequence Alignment/Map - BED : Browser Extensible Data - CRAM : compression algorithm - MRF : Mapped Read Format - VCF : Variant Call Format. |
|
7. Quelques exemples d'apports du séquençage a. Le génome de l'homme : "ENCODE" Septembre 2012 - Publication de 30 articles par le consortium ENCODE ("The Encyclopedia of DNA Elements") qui montrent notamment que :
On estime que l'exome humain (partie de l'ADN constituée par les exons) correspond à environ 20.000 gènes codant des protéines (exome : 180.000 exons - 1,5% du génome humain - 30 Mbases). Pour accéder aux articles de manière interactive : Nature ENCODE explorer |
| Evolution du nombre de gènes estimés dans le génome humain | |||
| Technique | Date | Nombre de gènes estimés | Hypothèses et commentaires |
| "Calcul" initial | 1990 | 100 000 | Avec l'hypothèse que la taille moyenne d'un gène = 30 kb |
| Ebauche de séquençage du génome | 1994 | 71 000 | Résultat biaisé par les régions riches en gènes ? |
| Ilôts CpG | 80 000 | Avec l'hypothèse que 66% des gènes humains ont de tels "ilôts" | |
| Analyse des EST | 1994 | 64 000 | Gènes ayant un homologue dans GenBank - Redondance des EST de 50% |
| Chromosome 22 | 1999 | 45 000 | Correction liée à la haute densité en gène de ce chromosome |
| Technique "Exofish" ("Exon Finding by Sequences Homology") | 2000 | 28 000 - 34 000 | Avec l'hypothèse que les régions codantes sont plus conservées que les régions non-codantes. Comparaison des génomes homme - poisson ("Tetraodon nigroviridis") |
| EST | 2000 | 35 000 120 000 |
Nombre de gènes Nombre de transcrits |
| Premier "brouillon" du génome | 2001 | 30 000 - 40 000 | Gènes connus + prédictions |
| Comparaison avec le génome de la souris | 2002 | 30 000 | Gènes connus + prédictions |
| Analyse du génome de l'homme en cours d'aboutissement | 2004 | 20 000 - 25 000 | Gènes connus + prédictions |
| 2007 | 20 000 | Annotation des gènes améliorée | |
| Analyse du génome de l'homme "aboutie" | 2012 | 20 687 gènes codant des protéines | "The ENCODE Project Consortium" Nature 489, 57-74 (2012) |
| Analyse conjointe du génome et du protéome (5 niveaux d'évidence d'existence des protéines) de l'homme | 2018 - 2020 | 19 823 gènes codant des protéines 20 399 protéines (dont 17 694 protéines PE1) |
HUPO (Human Proteome Project) NextProt (Swiss Institute of Bioinformatics) |
Publication du consortium Telomere-to-Telomere (T2T) |
2021 | 19 969 gènes codant des protéines 3,055 milliards de paires de bases |
Caractéristiques principales de la référence T2T-CHM13
|
| Chez presque tous les organismes (et donc chez l'homme), on découvre un nombre croissant important de microprotéines ou micropeptides codés par des ARN qui jusqu'à lors étaient annotés "non codant". Le nombre réel de protéines est donc sous-estimé. | |||
| Quelques données sur le génome humain (les chiffres ne sont évidemment pas définitifs, le décryptage complet n'étant pas terminé)* | |
| Régions transcrites en ARN : 0,05% | Tailles moyennes :
|
| Régions codant pour des protéines : 1,2% | |
| Introns : 31% | |
| ADN intergénique : 61% | |
| ADN satellite : 6 - 7% | |
| Pseudogènes : 1 - 1,2% | |
| Eléments transposables : 42 - 46 % | |
* 2015 : reconstruction du génome de 2504 individus issus de 26 populations ("The 1000 Genomes Project Consortium"). |
|
Les mutations somatiques dans les génomes de cellules cancéreuses sont causées par de multiples processus, chacun générant une signature de mutations caractéristique. Le consortium PCAWG ("Pan-Cancer Analysis of Whole Genomes") de l'ICGC ("International Cancer Genome Consortium") et du TCGA ("The Cancer Genome Atlas") a caractérisé les signatures de mutations la plupart des types de cancer de l'homme en analysant près de 85 millions de mutations somatiques dans 4645 génomes entiers et 19.184 séquences d'exomes. Cette analyse a permis d'identifier 49 substitutions de base unique, 11 substitutions de base en doublet, 4 substitutions de base en cluster et 17 petites signatures d'insertion et de suppression.
|
|
b. Evolution de ENCODE : "Comparative modENCODE/ENCODE" (analyse comparative de métazoaires) Les données de ENCODE permettent désormais la comparaison spatio-temporelle des génomes, des transcriptomes, des séquences qui codent les divers types d'ARN, de la structure de la chromatine, des interactions ADN-protéine ... C'est ce qui a été publié en 2014 pour 3 espèces de métazoaires apparemment distantes : l'homme, le ver nématode (Caenorhabditis elegans) et la mouche (Drosophila melanogaster).
L'ensemble des données (notamment le stockage des données de séquençage de milliards de fragments) est disponible à la sous-partie du consortium ENCODE : "Comparative modENCODE/ENCODE".
Araya et al. (2014) Nature 512, 400-405 : distribution spatio-temporelle dans le génome de Caenorhabditis elegans des sites de liaison de 92 facteurs de transcription et protéines régulatrices à plusieurs stades de développement (241 expériences ChIP-seq). |
c. Visualisation des protéines de Caenorhabditis elegans Des constructions génétiques permettent l'expression in vivo des protéines de Caenorhabditis elegans. Ces protéines sont marquées par affinité par des sondes fluorescentes (voir : Sarov et al., 2012). 73% des protéines sont ensuite actuellement visualisables in vivo dans chaque type de compartiment à un moment donné de l'existence de Caenorhabditis elegans (figure ci-dessous).
Source : TransgeneOme TransgeneOme : Une plateforme dédiée à Caenorhabditis elegans transgènique à l'échelle de son génome. Elle contient 16.000 constructions (fosmides) sur les 20.000 gènes codant des protéines. Remarque : ce ver nématode est à l'origine de la description exacte du phénomène d'interférence ARN (RNAi) par Fire et Mello. |
d. La domestication du chien Cette domestication a été un épisode important dans le développement de la civilisation humaine. Cependant, la période et les lieux de cet événement ne sont pas encore clairement établis. Par ailleurs, on connaît mal les changements génétiques qui ont accompagné la transformation des loups primitifs en chiens domestiques. Un re-séquençage du génome entier du chien et du loup a permis d'identifier 3,8 millions de variants génétiques utilisés. Ces variants ont a leur tour permis d'identifier 36 régions du génome, probablement cibles de la sélection au cours de la domestication du chien (Axelsson et al., 2013).
Il est ainsi fort probable que les processus adaptatifs qui ont permis aux ancêtres du chien moderne de prospérer avec une alimentation riche en amidon (issus principalement des déchets des humains sédentarisés), par rapport à l'alimentation carnivore du loup, a constitué une étape cruciale dans la domestication du chien. |
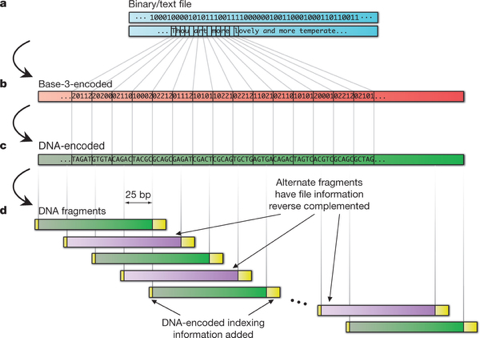
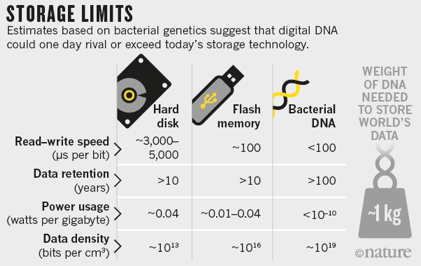
e. Le codage de l'information numérique dans de l'ADN La production de ressources numériques, la transmission de données et leur stockage ont révolutionné notre vie moderne. Cependant, de manière parallèle, les tâches d'archivage actif et d'entretien en continu des médias numériques sont de plus en plus complexes. La molécule d'ADN s'avère un support particulièrement attractif pour le stockage de l'information. C'est, peut-être, le support de l'avenir, du fait notamment de ses capacités d'encodage à haute densité de l'information et de sa longévité dans des conditions de conservation faciles à mettre en oeuvre. Récemment, un éventail de formats de fichiers courants en informatique ont été codés sous la forme d'ADN (Goldman et al., 2013):
Soit un total de codage dans une molécule d'ADN de l'équivalent de 739 kilo-octets de stockage sur un disque dur avec un taux estimé d'informations de Shannon de 5.2 106 bits.
L'ADN a été synthétisé, puis séquencé et les fichiers d'origine ont été reconstruits avec une précision de 100%. L'analyse théorique indique que le stockage dans de l'ADN est :
|
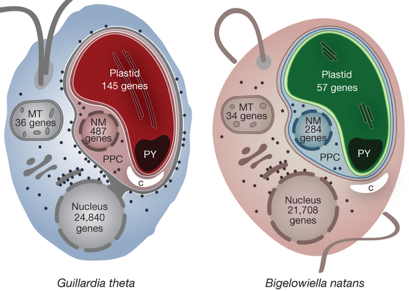
f. Support de la théorie endosymbiotique Les génomes nucléaires de 2 algues unicellulaires, remarquables par leur complexité génétique et cellulaire, ont été séquencés : la cryptophyte Guillardia theta et la chlorarachniophyte Bigelowiella natans. Le transfert de gènes endosymbiotiques, c'est-à-dire le mouvement de l'ADN de l'endosymbiote vers l'hôte avant, pendant et après l'évolution d'un organite, a eu un rôle notable dans l'évolution des algues et de leurs génomes nucléaires. Les plastes secondaires de ces algues (qui ont évolué indépendamment) sont uniques du fait qu'ils ont conservé un noyau "relique" de l'endosymbiote, appelé nucléomorphe. Les cellules des cryptophytes et des chlorarachniophytes ont ainsi 4 génomes et contiennent des systèmes sub-cellulaires complexes pour l'adressage des protéines et pour la coordination entre les compartiments. Les algues Guillardia theta et Bigelowiella natans ont des plastes entourés par 4 membranes (figure ci-dessous).
Source : Curtis et al. (2012) Chez les cryptophytes, la membrane ultra-périphérique est en continuité de l'enveloppe nucléaire et sa surface est parsemée de ribosomes. Entre les paires de membranes internes et externes se trouve le compartiment péri-plastidial (PPC), qui contient le nucléomorphe (NM). Les chiffres indiquent le nombre estimé de gènes codant des protéines dans les génomes des plastes, des mitochondries (MT), du nucléomorphe et du noyau. C : hydrates de carbone; PY : pyrénoïdes. |
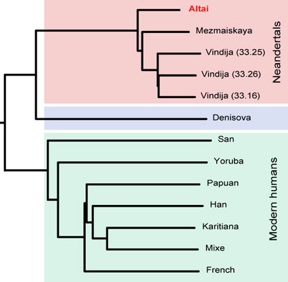
g. Les origines de l'espèce humaine : le séquençage du génome de l'homme de Neanderthal Le premier brouillon du génome de l'homme de Neanderthal a été publié en 2010 par l'équipe de Svante Pääbo (Green et al. (2010) "A Draft Sequence of the Neandertal Genome" Science 328, 710 - 722). En mars 2013, la même équipe du "Max Planck Institute for Evolutionary Anthropology" (Leipzig - Allemagne) a publié une séquence encore plus précise de ce génome. Il est issu d'un ADN extrait d'un os d'orteil découvert dans la grotte de Denisova en Sibérie du Sud en 2010.
Source : Neanderthal genome project (2013) Le séquençage a été effectué avec une plate-forme Illumina HiSeq : (i) il correspond à à une couverture moyenne du génome 50 fois supérieure à celle du brouillon de 2010; (ii) 99,9 % des 1.7 Gb des séquences d'ADN cartographiables de façon unique sont couvertes au moins 10 fois. La conclusion phare est qu'il semblerait que, contrairement à ce que pensaient de nombreux chercheurs, des Néandertaliens et des hommes modernes se soient mélangés par le passé. Cette conclusion est diamètralement opposée à celle formulée par la même équipe en 1997 qui était que "l'homme de Neandertal n'aurait pas contribué à notre patrimoine génétique et constitue une espèce distincte de la nôtre, sans métissage possible". Il est à noter que le premier génome séquencé était celui de la mitochondrie, bien plus petit et donc moins riche en informations. |
| Outils pour l'analyse de l'ADN ancien (fossile) | ||
| Logiciel | Lien | Description |
| CASCADE | Available by contacting authors | LIMS for aDNA experimental workflow |
| PALEOMIX | https://paleomix.readthedocs.io/en/latest/ | Read alignment and processing, phylogenomics |
| nf-core/EAGER |
https://eager.readthedocs.io/en/latest/index.html https://github.com/nf-core/eager |
Read alignment and processing |
| mapDamage2 | https://ginolhac.github.io/mapDamage | Post-mortem DNA damage assessment |
| PMDtools | https://github.com/pontussk/PMDtools | Selection of reads showing signatures of post-mortem DNA damage |
| Schmutzi | https://grenaud.github.io/schmutzi/ | Contamination estimates based on mitochondrial DNA data |
| DICE | https://github.com/grenaud/dice | Contamination estimates based on nuclear data |
| VerifyBamID | https://github.com/statgen/verifyBamID/releases | Identification of contamination and/or sample swaps |
| Gargammel | https://grenaud.github.io/gargammel/ | aDNA read simulator |
| metaBIT | https://bitbucket.org/Glouvel/metabit/src/master/ | Taxonomic profiling of (ancient) metagenomic data |
| HOPS | https://github.com/rhuebler/HOPS | Taxonomic profiling of (ancient) metagenomic data |
| MEx-IPA | https://github.com/jfy133/MEx-IPA | Interactive viewer of MALT taxonomic assignments |
| coproID | https://github.com/nf-core/coproid | Identification of the host sources of faecal material |
| epiPALEOMIX | https://bitbucket.org/khanghoj/epipaleomix/wiki/Home | Inference of aDNA methylation and nucleosome mapping |
| DamMet | https://github.com/KHanghoj/DamMet | Inference of aDNA methylation, accounting for DNA damage, sequencing and genotyping errors |
| ANGSD | http://www.popgen.dk/angsd/index.php/ANGSD | Variant identification, population genetics inference |
| ATLAS | https://bitbucket.org/wegmannlab/atlas/wiki/Home | Variant identification |
| ADMIXtools | https://github.com/DReichLab/AdmixTools | Population genetics inference |
| smartPCA | https://github.com/chrchang/eigensoft/wiki/smartpca | PCA and Procrustes PCA projection |
| bammds | https://savannah.nongnu.org/projects/bammds/ | Multidimensional scaling |
| PCAngsd | http://www.popgen.dk/software/index.php/PCAngsd | PCA, admixture and selection signatures |
| DATES | https://github.com/priyamoorjani/DATES | Inference of admixture timing |
| LSD | https://bitbucket.org/plibrado/LSD/src | Selection signatures |
| GRoSS | https://github.com/FerRacimo/GRoSS | Selection signatures |
| ROHan | http://grenaud.github.io/ROHan/ | Heterozygosity estimates and runs of homozygosity |
| hapROH | https://pypi.org/project/hapROH/ | Inbreeding inference from low-coverage data |
| lcMLkin | https://github.com/COMBINE-lab/maximum-likelihood-relatedness-estimation | Kinship inference |
| READ | https://bitbucket.org/tguenther/read/src/master/ | Kinship inference |
| SourceTracker | https://github.com/danknights/sourcetracker | Metagenomic authentication |
| Source : Orlando et al. (2021) "Ancient DNA analysis" Nat. Rev. Meth. Primers 1, Art. number 14 | ||
| Quelques articles clé dans l'histoire du séquençage de l'ADN ancien | |
| Article | Description |
| Poinar et al. (1998) Sciences 281, 402 - 406 | Première application du séquençage d'ADN de nouvelle génération à des spécimens anciens (coproscopie moléculaire). Description de la nature métagénomique des vestiges paléontologiques ainsi que des données à l'échelle de la mégabase du génome du mammouth laineux. |
| Willerslev et al. (2003) Science 300, 791 - 795 | Première analyse de l'ADN environnemental avant l'avènement du séquençage de l'ADN de nouvelle génération et établit la persistance à long terme de l'ADN des paléocommunautés dans les holocènes et pléistocènes. |
| Briggs et al. (2007) Proc. Natl Acad. Sci. 104, 14616 - 14621 | Premier modèle statistique de dégradation post-mortem de l'ADN génomique de Néandertal. Impact sur les modèles de mésincorporation de nucléotides (base de critères importants pour l'authentification des données). |
| Rasmussen et al. (2010) Nature 463, 757 - 762 | Premier génome humain séquencé à partir de la tige pilaire d'un Paléo-Inuit âgé de 4.000 ans. Preuves d'une discontinuité génétique avec les Groenlandais modernes, soutenant de multiples vagues de migration vers le Groenland arctique. |
| Green et al. (2010) Sciences 328, 710 - 722 | Premier génome de Néandertal obtenu à partir d'extraits d'ADN de 3 os paléontologiques, établissant l'héritage génétique de Néandertal au sein des génomes humains modernes et décrivant d'importantes méthodologies au laboratoire qui ont façonné la décennie suivante de recherche sur l'ADN. |
| Reich et al. (2010) Nature 468, 1053 - 1060 | Découverte des Denisoviens, lignée jusque-là inconnue d'hominidés archaïques qui vivaient dans le sud de la Sibérie il y a au moins 50.000 ans. Première description d'un représentant de l'arbre évolutif Homo à partir de données moléculaires et en absence de restes macro-fossiles présentant des caractéristiques morphologiques claires. |
| Bos et al. (2011) Nature 478, 506 - 510 | Premier génome complet d'un ancien agent pathogène bactérien (Yersinia pestis) à partir d'os humains d'individus morts de la peste noire (1347-1348). |
| Meyer et al. (2012) Sciences 338, 222 - 226 | Premier génome de haute qualité d'un hominidé archaïque et première procédure expérimentale pour la préparation d'une banque d'ADN à partir de matrices d'ADN simple brin. L'approche surpasse les autres technologies de l'époque en termes de sensibilité et de complexité et minimise la perte de molécules d'ADN authentiques. |
| Orlando et al. (2013) Nature 499, 74 - 78 | Plus ancien génome séquencé à ce jour, d'un métapode de cheval conservé dans le pergélisol (âge 780.000 à 560.000 ans). |
| Fu et al. (2013) Proc. Natl Acad. Sci. 110, 2223 - 2227 | Première application de l'enrichissement de cibles en solution à l'échelle du génome. Analyse de la séquence d'un chromosome complet d'un humain anatomiquement moderne de 40.000 ans de Chine (grotte de Tianyuan). |
| Dabney et al. (2013) Proc. Natl Acad. Sci. 110, 15758 - 15763 | Nouvelle méthode d'extraction d'ADN à partir de restes osseux anciens adaptée à la nature ultracourte et largement endommagée des molécules d'ancien ADN. Cette méthodologie a permis de récupérer des séquences complètes du génome mitochondrial de spécimens d'ours des cavernes vieux de 300.000 ans (Pléistocène moyen) à Atapuerca (Espagne). |
| Warinner et al. (2014) Nat. Genet. 46, 336 - 344 | Première analyse métagénomique et paléoprotéomique de la plaque dentaire ancienne. Démontration de la préservation des signatures microbiennes orales, du contenu alimentaire et de marqueurs d'inflammation. |
| Pedersen et al. (2014) Genome Res. 24, 454 - 466 | Premier rapport d'épigénomes anciens (Paléo-Inuitde 4.000 ans), tirant parti des signatures de dégradation de l'ADN post-mortem pour déduire statistiquement la méthylation de l'ADN et le positionnement des nucléosomes. |
| Rohland et al. (2015) Philos. Trans. R. Soc. Londres. B Biol. Sci. 370, 20130624 | Première méthode (traitement partiel à l'uracile-ADN-glycosylase) de préparation de bibliothèques d'ADN compatible avec une automatisation complète. Description de diverses approches pour authentifier les données tout en minimisant l'impact de la mauvaise incorporation d'ADN post-mortem sur les analyses subséquentes. |
| ADN ancien : fragments d'ADN ultracourts et dégradés qui sont conservés dans des matériaux subfossiles, y compris dans les tissus durs (exemples : os, dents, coquillages) et dans les tissus mous (exemple : peau momifiée, cheveux momifiés) et dans les sédiments. Voir la carte des sites de fouilles et de découvertes : "Ancient Human DNA uMap" Lessivage stratigraphique : migration de l'ADN au travers des strates de sédiments causée par le mouvement de l'eau, la croissance de micro-organismes ou la bioturbation. Elle compromet la fiabilité de la stratigraphie : l'ordre, la position et l'âge des couches géologiques formées par les différents amas de sédiments. |
|
| Source : Orlando et al. (2021) | |
h. Reconstitution de la domestication des agrumes comestibles La production des agrumes comestibles représentait 9 milliards de dollars en 2012. Les premiers agrumes comestibles ont été cultivés il y a plusieurs milliers d'années en asie du sud-est mais les voies qu'ont suivies les différentes variétés cultivées actuellement ont été perdues. La très faible diversité génétique des agrumes comestibles les rend très vulnérables à diverses maladies. Le séquençage de plusieurs génomes de mandarine/clémentine, d'orange et de pamplemousse et la comparaison de ces génomes (synténie - ancêtre eudicotyledon hexaploïde) permettent d'établir des stratégies pour améliorer la résistance des agrumes modernes (Wu et al. (2014) Nature Biotech. 32, 656-62). - Phytozome v.10 : "High-quality reference genome from a haploid derivative of Clementine mandarin (C. x clementina cv. Clemenules)" |
8. Etude des éléments de la régulation de la transcription - Structure de la chromatine et épigénomique Chromatine = ADN + protéine (histones et non-histone) + ARN. Les chromosomes en métaphase représentent le degré le plus élevé de compaction de la chromatine. Voir un cours sur l'épigénétique (modifications de l'ADN et des histones). L'accessibilité des protéines (facteurs de transcription et de régulation de la transcription) dépend de la compacité de la chromatine. Le contrôle de la structure de la chromatine est donc un autre mode de contrôle de la transcription des gènes. De très nombreuses technologies sont développées pour :
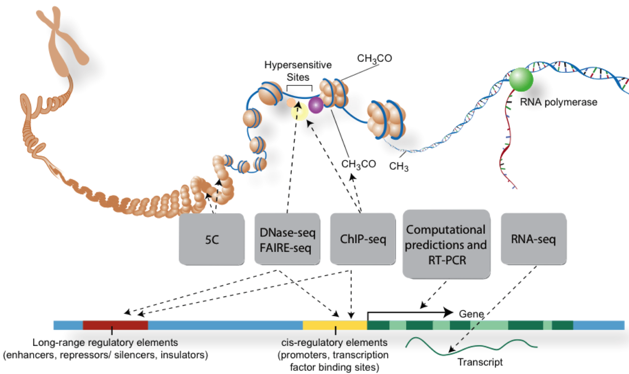
Figure ci-dessous : Techniques de traitement des acides nucléiques avant séquençage pour l'analyse de parties spécifiques des génomes.
Source : ENCODE Par exemple :
|
| Eléments du génome cartographiées | Techniques utilisées |
| Régions transcrites en ARN | RNA-seq / CAGE / RNA-PET / annotation manuelle |
| Régions codant des protéines | Spectromètrie de masse |
| Sites de fixation des facteurs de transcription | ChIP-seq / DNase-seq |
| Structure de la chromatine | DNase-seq / FAIRE-seq / Histone ChIP-seq / MNase-seq |
| Sites de méthylation de l'ADN | RRBS |
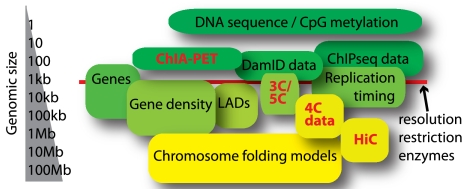
Le développement de nouvelles technologies permet l'étude du chromosome interactome et des interactions chromatine-chromatine à longue distance in vivo :
|
Source : de Wit & de Laat (2012) |
Définitions des acronymes de ces nouvelles technologies
ChIPBase : base de données et plate-forme pour le décodage des cartes de liaison, des facteurs de transcription, des profils d'expression, de la régulation de la transcription de longs ARN non codants ("long non-coding RNAs" : lncRNAs, lincRNAs), de microRNA et autres ARN non codant (snoRNAs, tRNAs, snRNAs, ...) et des gènes codant des protéines. |
|
| Appellations et acronymes de techniques de séquençage issues des NGS | |
| RNA Transcription | RNA Structure |
| Chromatin Isolation by RNA Purification (ChIRP-Seq) Global Run-on Sequencing (GRO-Seq) Ribosome Profiling Sequencing (Ribo-Seq)/ARTseq™ RNA Immunoprecipitation Sequencing (RIP-Seq) High-Throughput Sequencing of CLIP cDNA library (HITS-CLIP) Crosslinking and Immunoprecipitation Sequencing (CLIP-Seq) Photoactivatable Ribonucleoside–Enhanced Crosslinking and Immunoprecipitation (PAR-CLIP) Individual Nucleotide Resolution CLIP (iCLIP) Native Elongating Transcript Sequencing (NET-Seq) Targeted Purification of Polysomal mRNA (TRAP-Seq) Crosslinking, Ligation, and Sequencing of Hybrids (CLASH-Seq) Parallel Analysis of RNA Ends Sequencing (PARE-Seq) Genome-Wide Mapping of Uncapped Transcripts (GMUCT) Transcript Isoform Sequencing (TIF-Seq) Paired-End Analysis of TSSs (PEAT) |
Selective 2'-Hydroxyl Acylation Analyzed by Primer Extension Sequencing (SHAPE-Seq) Parallel Analysis of RNA Structure (PARS-Seq) Fragmentation Sequencing (FRAG-Seq) CXXC Affinity Purification Sequencing (CAP-Seq) Alkaline Phosphatase, Calf Intestine-Tobacco Acid Pyrophosphatase Sequencing (CIP-TAP) Inosine Chemical Erasing Sequencing (ICE) m6A-Specific Methylated RNA Immunoprecipitation Sequencing (MeRIP-Seq) |
| Low-Level RNA Detection | Low-Level DNA Detection |
| Digital RNA Sequencing Whole-Transcript Amplification for Single Cells (Quartz-Seq) Designed Primer–Based RNA Sequencing (DP-Seq) Switch Mechanism at the 5' End of RNA Templates (Smart-Seq) Unique Molecular Identifiers (UMI) Cell Expression by Linear Amplification Sequencing (CEL-Seq) Single-Cell Tagged Reverse Transcription Sequencing (STRT-Seq) |
Single-Molecule Molecular Inversion Probes (smMIP) Multiple Displacement Amplification (MDA) Multiple Annealing and Looping–Based Amplification Cycles (MALBAC) Oligonucleotide-Selective Sequencing (OS-Seq) Duplex Sequencing (Duplex-Seq) |
| DNA Methylation | DNA-Protein Interactions |
| Bisulfite Sequencing (BS-Seq) Post-Bisulfite Adapter Tagging (PBAT) Tagmentation-Based Whole Genome Bisulfite Sequencing (T-WGBS) Oxidative Bisulfite Sequencing (oxBS-Seq) Tet-Assisted Bisulfite Sequencing (TAB-Seq) Methylated DNA Immunoprecipitation Sequencing (MeDIP-Seq) Methylation-Capture (MethylCap) Sequencing Methyl-Binding-Domain–Capture (MBDCap) Sequencing 79 Reduced-Representation Bisulfite Sequencing (RRBS-Seq) |
DNase l Hypersensitive Sites Sequencing (DNase-Seq) MNase-Assisted Isolation of Nucleosomes Sequencing (MAINE-Seq) Chromatin Immunoprecipitation Sequencing (ChIP-Seq) Formaldehyde-Assisted Isolation of Regulatory Elements (FAIRE-Seq) Assay for Transposase-Accessible Chromatin Sequencing (ATAC-Seq) Chromatin Interaction Analysis by Paired-End Tag Sequencing (ChIA-PET) Chromatin Conformation Capture (Hi-C/3C-Seq) Circular Chromatin Conformation Capture (4-C or 4C-Seq) Chromatin Conformation Capture Carbon Copy (5-C) |
| Source : Illumina - publications-reviews | |
10. Séquençage du transcriptome : méthode "RNA-seq" C'est une technologie récente et assez complexe. Des centaines de millions de fragments trés courts (quelques dizaines de nucléotides) sont générés et séquencés ("ultra high-throughput short reads"). Quelques avantages de la méthode "RNA-seq"
Cette technique permet :
Source : Buckingham (2003)
Source : Amin et al. (2019) Cette technique procure des informations issues de la comparaison avec des génomes complets. Il n'est pas nécessaire d'avoir des connaissances sur le génome étudié. Cependant, si l'on dispose de génomes de "référence", c'est une méthode de choix pour améliorer sensiblement leur annotation.
Les difficultés d'assemblage des lectures et de reconstruction des transcrits pleine longueur sont les suivantes (liste non exhaustive) :
Les nouvelles technologies de séquençage ont permis d'établir que la méthylation du transcriptome (épitranscriptome) est un processus quasi général au même titre que la méthylation du génome (épigénome). |
| a. Méthodes pour l'assemblage des lectures |
| Les approches de type "Mapping-first" | Les approches de type "Assembly-first (de novo)" | |
| Elles alignent d'abord toutes les lectures par rapport à un génome de référence (non annoté) puis assemblent les séquences (alignements de séquences chevauchantes) et enfin essayent de couvrir les bordures d'épissage alternatif avec les lectures de type "single-end sequencing / paired-end sequencing". | Elles utilisent les lectures pour assembler les séquences des transcrits qui seront ensuite positionnés sur un génome de référence (s'il en existe un). | |
| Ces approches ont, en principe, une sensibilité maximale mais dépendent de l'alignement correct [lectures - génome de référence] qui est compliqué du fait de l'épissage alternatif, des erreurs de séquençage et de l'absence de génomes de référence dans de nombreux cas. | Ces approches ne nécessitent pas d'alignement [lectures - génome de référence], point capital si on ne dispose pas de génome de référence, ou si celui-ci contient beaucoup de gap ou s'il est très fragmenté ou s'il est modifié de façon substantielle. | |
| Exemples de logiciels : |
Exemples de logiciels :
|
|
Autres moyens bioinformatiques
Voir une liste très complète des ressources logicielles pour l'analyse des données RNA-seq. 3 types de formats de fichiers sont couramment utilisés :
|
||
| La qualité des assemblages | Définition de RPKM | |
Elle est mesurée par la taille et la précision de leurs contigs. La taille d'un assemblage est corrélée à des valeurs statistiques : la longueur maximale, la longueur moyenne, la longueur totale combinée et la valeur du contig N50. Le contig N50 est la longueur du plus petit contig dans l'ensemble qui contient le moins de contigs (donc l'ensemble qui contient les contigs les plus grands) et dont la longueur combinée de ces contigs représente au moins 50% de l'assemblage. |
RPKM : "Reads Per Kilobase of exon model per Million fragments mapped" = C . 109 / [N x L] avec :
Mortazavi et al. (2008) "Mapping and quantifying mammalian transcriptomes by RNA-Seq" Nat. Meth. 5, 621-628 |
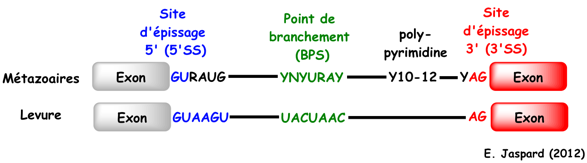
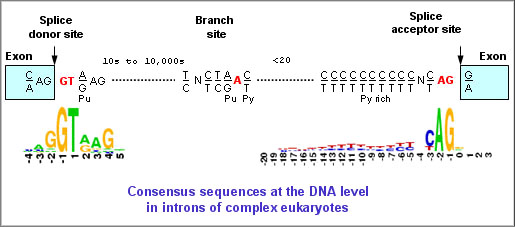
b. Caractéristiques des bordures exon-intron ("exon-intron borders") Voir un cours sur l'épissage des ARN messager et le spliceosome. La plupart des introns commencent par la séquence consensus 5'-GU et finissent par la séquence consensus AG-3'. Ces séquences sont appelés respectivement "site donneur lors de l'épissage" et "site accepteur lors de l'épissage" ("splice donor site" et "splice acceptor site"). Il existe en général une région riche en nucléotides pyrimidiques (C et U) en amont du site AG. Figure ci-dessous : Séquences consensus de pré-ARNm subissant un épissage.
Légende : BPS : "branch point sequence"; 5'SS : "5' splicing site"; 3'SS : "3' splicing site"; N : n'importe quel nucléotide; R : une purine; Y : une pyrimidine
Dans 60% des cas, l'extrémité de la séquence de l'exon situé en 5' (site donneur) est (A/C)AG et l'extrémité de la séquence de l'exon situé en 3' (site accepteur) est G (voire A).
Source : "RNA sequence analysis tools" |
| c. Méthodes pour la reconstruction des transcrits |
|
Les logiciels d'alignement de lectures dans des régions non épissées ("unspliced reads aligner"). Ils sont utilisés pour aligner les lectures avec un transcriptome de référence ou un génome de référence. |
Les logiciels d'alignement de lectures dans des régions épissées ("spliced reads aligner"). Ils sont utilisés pour aligner les lectures avec un génome de référence. Ces lectures peuvent enjamber des introns et nécessitent l'introduction de grands gaps. |
|
Il existe 2 principaux types de méthodes : - la méthode "seed" (exemple de logiciel : Stampy) : aligne de courtes sous-séquences ("seed" - points d'ancrage) de chaque lecture avec une référence, nécessitant une concordance parfaite ("perfect matches") de tous les nucléotides de ces sous-séquences. - la méthode Burrows-Wheeler (exemple de logiciel : Bowtie) : elle réorganise les caractères dans une séquence, permettant une meilleure compression des données et ainsi d'utiliser moins de capacité mémoire lors de l'alignement des lectures sur un génome. La méthode Burrows-Wheeler crée un index de la séquence de référence et recherche des correspondances parfaites. Les discordances ("mismatches") sont autorisées mais s'accompagnent d'une augmentation exponentielle de la complexité du calcul. La méthode Burrows-Wheeler est plus rapide mais moins sensible que la méthode "seed". |
Les logiciels de ce type placent les lectures qui enjambent les jonctions d'épissage en les fractionnant en segments plus petits. Puis ils déterminent la meilleure correspondance (théorique) sur la base de scores d'alignement et de signaux consensus d'épissage (di-nucléotides consensus 5'-GU et AG-3'). Il existe 2 principaux types de méthodes : - la méthode "exon-first" (exemple : logiciel TopHat) : elle cartographie l'ensemble des lectures sur le génome avec une méthode du type "unspliced read aligners", puis elle recherche des alignements dans les zones d'épissage avec les lectures non cartographiées. - la méthode "seed-and-extend" (exemple : logiciel GSNAP) : c'est une stratégie d'alignement qui construit d'abord une table de hachage contenant l'emplacement de chaque k-mer ("seed" - point d'ancrage) sur le génome de référence. Ces algorithmes étendent ensuite ces point d'ancrage dans les deux directions pour trouver le ou les meilleur(s) alignement(s) pour chaque lecture. La méthode "seed-and-extend" est plus lente mais plus sensible. |
|
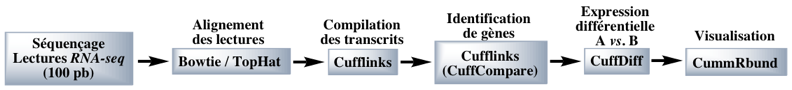
Figure ci-dessous : exemple de suite logicielle ("pipeline") pour l'analyse RNA-seq.
Voir une liste (impressionnante) de logiciels dédiés à l'analyse des résultats RNA-seq. Principe (très simplifié) de la reconstruction des transcrits avec des graphes de type de Bruijn Dans ce type de graphe, un nœud est défini par une séquence de nucléotides d'une longueur k fixe (appelée "k-mer"). Cette longueur k est beaucoup plus courte que la longueur d'une lecture. Les noeuds sont reliés par des arêtes si les noeuds se chevauchent parfaitement sur (k-1) nucléotides. Chaque nœud est relié à un nœud "jumeau" qui est la série inverse des séquences complémentaires des "k-mer". Cela permet de prendre en compte le chevauchement entre les lectures de brins opposés. Cette représentation compacte permet d'énumérer toutes les solutions par lesquelles les séquences des transcrits peuvent être reconstruites : pour l'assemblage du transcriptome, chaque chemin dans le graphe représente un transcrit possible. Avenir des méthodes d'assemblages de courts fragments
|
| 11. Liens Internet et références bibliographiques |
| "Précis de génomique" - Gibson & Muse (2004) - Ed. De Boeck Université - ISBN : 2-8041-4334-1 | |
|
Méthode de séquençage de F. Sanger Sanger et al. (1977) "DNA sequencing with chain-terminating inhibitors" Proc. Natl Acad. Sci. USA 74, 5463 - 5467 Film (format QuickTime) : "Dideoxy Sequencing of DNA" Voir l'animation : "Sanger sequencing" |
|
|
ENCODE ENCODE : The Encyclopedia of DNA Elements |
|
Le séquençage des génomes - Université Jussieu Génoscope : Questions fréquemment posées à propos du génome humain. "DNA Sequencing Costs : Data from the NHGRI Large-Scale Genome Sequencing Program" "Transcriptome Shotgun Assembly (TSA) Database" DNAmod : base de données de modifications chimiques de l'ADN RMbase : base de données de séquençage d'épitranscriptomes - analyse des modifications post-transcriptionnelles des ARN |
|
|
Quelques articles en relation avec les nouvelles technologies de séquençage
|
|
La méthode "Massively parallel signature sequencing" - MPSS - Brenner et al. (2000) Une séquence signature de 16 à 20 pb (en moyenne 17 pb) fixée à une bille est séquencée / identifiée. Cette identification est effectuée en parallèle sur des centaines de milliers de billes et environ 1 million de signatures sont obtenues par expérience. Voir une animation décrivant cette technique. Caractéristiques de cette technique :
Application à Arabidopsis
|
|
|
Smith et al. (1986) "Fluorescence detection in automated DNA sequencing" Nature 321, 674 - 679 Ronaghi et al. (1998) "A sequencing method based on real-time pyrophosphate" Science 281, 363 - 365 Ahmadian et al. (2006) "Pyrosequencing: History, biochemistry and future" Clinica Chimica Acta 363, 83 - 94 Edwards & Batley (2010) "Plant genome sequencing: applications for crop improvement" Plant Biotechnol. J. 8, 2 - 9 Sboner et al. (2011) "The real cost of sequencing: higher than you think" Genome Biology 12, 125 |
|
|
Wang et al. (2012) "TILLING in extremis" Plant Biotechnol. J. 10, 761 - 772 Sarov et al. (2012) "A Genome-Scale Resource for In Vivo Tag-Based Protein Function Exploration in C. elegans" Cell 150, 855 - 866 Lu et al. (2012) "Effective driving force applied on DNA inside a solid-state nanopore" Phys. Rev. E 86, 01192-1 - 01192-8 Vlassarev & Golovchenko (2012) "Trapping DNA near a Solid-State Nanopore" Biophysical J. 103, 352 - 356 |
|
|
Curtis et al. (2012) "Algal genomes reveal evolutionary mosaicism and the fate of nucleomorphs" Nature 492, 59 - 65 Axelsson et al. (2013) "The genomic signature of dog domestication reveals adaptation to a starch-rich diet" Nature 495, 360–364 Goldman et al. (2013) "Towards practical, high-capacity, low-maintenance information storage in synthesized DNA" Nature 494, 77 - 80 Green et al. (2010) "A Draft Sequence of the Neandertal Genome" Science 328, 710 - 722 |
|
Bock et al. (2010) "Genome-wide mapping of DNA methylation: a quantitative technology comparison" Nat. Biotechnol. 28, 1106 - 1114 Manrao et al. (2012) "Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase" Nat. Biotechnol. 30, 349 - 353 Shendure & Aiden (2012) "The expanding scope of DNA sequencing" Nature Biotech. 30, 1084–1094 |
|
Ramaswami et al. (2013) "Identifying RNA editing sites using RNA sequencing data alone" Nature Meth. 10, 128 - 13 Hoque et al. (2013) "Analysis of alternative cleavage and polyadenylation by 3' region extraction and deep sequencing" Nature Meth. 10, 133 - 139 Benjamin et al. (2014) "Comparing reference-based RNA-Seq mapping methods for non-human primate data" BMC Genomics 15, 570 |
|
Quick et al. (2016) "Real-time, portable genome sequencing for Ebola surveillance" Nature 530, 228 - 232 Lek et al. (2016) "Analysis of protein-coding genetic variation in 60,706 humans" Nature 536, 285 291 Shendure et al. (2017) "DNA sequencing at 40: past, present and future" Nature 550, 345 - 353 |
|
Jain et al. (2018) "Nanopore sequencing and assembly of a human genome with ultra-long reads" Nat. Biotechnol. 36, 338 - 345 Omenn et al. (2018) "Progress on Identifying and Characterizing the Human Proteome: 2018 Metrics from the HUPO Human Proteome Project" J. Proteome Res. 17, 4031 - 4041 Noakes et al. (2019) "Increasing the accuracy of nanopore DNA sequencing using a time-varying cross membrane voltage" Nat. Biotechnol. 37, 651 - 656 Amin et al. (2019) "Evaluation of deep learning in non-coding RNA classification" Nat. Machine Intell. 1 246 - 256 |
|
Zahn-Zabal et al. (2020) "The neXtProt knowledgebase in 2020: data, tools and usability improvements" Nucleic Acids Res. 48, D328 - D334 Carr et al. (2020) "Nanopore Sequencing at Mars, Europa and Microgravity Conditions" NPJ Microgravity 6, 24 ICGC/TCGA PCAWG Consortium (2020) "Pan-cancer analysis of whole genomes" Nature 578, 82 - 93 |
|
Nurk et al. (2021) "The complete sequence of a human genome" biorxiv Orlando et al. (2021) "Ancient DNA analysis" Nat. Rev. Meth. Primers 1, Art. number 14 Wang et al. (2021) "Nanopore sequencing technology, bioinformatics and applications" Nat. Biotechnol. 39, 1348 - 1365 Lucas & Novoa (2023) "Long-read sequencing in the era of epigenomics and epitranscriptomics" Nat. Methods 20, 25 - 29 |
|
![]()





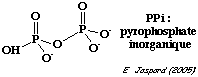




{kind=link}