
| Génomique fonctionnelle végétale - Arabidopsis thaliana et les plantes modèles |
| Tweet |
|
|
1. Rappels et définitions 2. Pourquoi Arabidopsis thaliana comme plante modèle ? 3. Le génome d'Arabidopsis thaliana 4. Bases de données relatives à Arabidopsis thaliana
|
5. Données sur le riz (Oryza sativa) 6. Medicago truncatula et le soja 7. Liens Internet et références bibliographiques |
1. Rappels et définitions - Sources principales : Unige-Ch / Wikipédia / P. Luchetta et al. - 2005 a. Principaux types de gènes
|
|
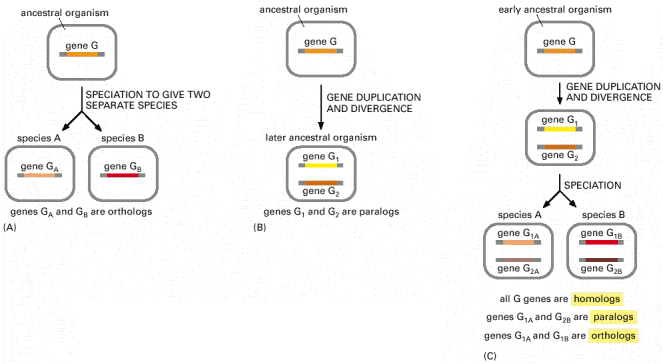
b. Gènes paralogues et gènes orthologues La duplication génique correspond à la multiplication de matériel génétique sur un chromosome. Des mutations peuvent alors affecter chaque copie des gènes après la duplication pour aboutir à une différenciation de deux gènes "frères" ou gènes paralogues. Donc, deux gènes au sein d'une espèce sont dits paralogues s'ils résultent d'une duplication génique. Lorsqu'une population évolue de manière indépendante (exemple : séparation géographique), ses caractéristiques génétiques évoluent de façon différente. Ce phénomène est appelé spéciation et il aboutit à la création de gènes orthologues : les mêmes gènes dans des espèces différentes. Donc deux gènes homologues (qui ont une forte similitude de séquence) de deux espèces différentes sont dits orthologues s'ils descendent d'un gène unique présent dans le dernier ancêtre commun aux deux espèces. Les espèces actuelles contiennent dans leur génome des gènes hérités d'un ancêtre commun (gènes homologues). Pour obtenir un arbre phylogénétique, on compare les gènes homologues. Quant un gène appartient à une famille multigénique, il est difficile de différencier les spéciations et les duplications. L'analyse des gènes paralogues est donc un élément important pour l'étude de l'évolution des génomes. Exemple de 2 types d'homologie de gènes basées sur des évènements évolutionnaires différents.
Source : "Molecular biology of the cell" (2002) Albert et al. (A) et (B) représentent les possibilités les plus simples. (C) représente un cas de figure plus complexe. |
|
c. Pseudogènes Ce sont des gènes non fonctionnels qui ont des similitudes avec un ou plusieurs gènes fonctionnels paralogues (gènes dupliqués au sein d'une même espèce). Ce sont donc des copies inactives de gènes fonctionnels. Cette inactivité est souvent due à l'absence de promoteur ou d'éléments de régulation. Les pseudogènes sont libres de toute contrainte sélective d'où une accumulation de mutations diverses dans leurs parties codantes ou non codantes. Il existe différents types de pseudogènes, notamment les pseudogènes dupliqués et les rétro-pseudogènes qui diffèrent par leur mécanisme d'apparition et leur mode d'évolution.
Les pseudogènes sont des indices d'évolution d'un organisme au cours du temps. Plus un organisme a évolué, plus grand est son nombre de pseudogènes. |
|
d. Rétroséquence Séquences qui résultent d'une transcription inverse puis qui sont intégrées au génome mais qui n'ont plus la capacité de se transposer. Elles ont certaines caractéristiques :
Les rétroséquences sont soit fonctionnelles (rétrogènes), soit non- fonctionnelles (rétro-pseudogènes). |
|
e. Rétrogène Le maintien de la fonction de ces séquences est un cas assez rare pour les raisons suivantes :
En conséquence, la majeure partie des rétroséquences sont des rétro-pseudogènes. |
| f. Eléments
transposables
Ceux sont des séquences d'ADN capable de se déplacer et de se multiplier de manière autonome dans un génome, par un mécanisme appelé transposition. Les transposons ont été identifiés par Barbara McClintock qui a étudié des mutations instables induites par ces éléments chez le maïs. Présents chez tous les organismes vivants, les éléments transposables sont un des constituants les plus importants des génomes eucaryotes. Exemples : 45% du génome de l'homme et plus de 70% chez le maïs. Les éléments transposables peuvent expliquer d'importantes différences dans les tailles du génome d'organismes. Les éléments transposables sont divisés en 2 classes selon leur mode de transposition : Ceux de classe I ou rétrotransposons : ils transposent via un intermédiaire ARN selon un mode réplicatif (copier - coller).
Leur ARN messager est rétro-transcrit en ADNc qui est intégré dans le génome. Les rétrotransposons sont divisés en 2 groupes :
Ceux de classe II ou transposons : ils transposent selon un mode conservatif (couper - coller). Ils sont caractérisés par la présence à leurs extrémités de séquences terminales inversement répétées (TIR). Ils sont de taille variable (100 à 20 000 pb).
Source : "La génomique végétale et les plantes cultivées" - Michel Caboche |
| Eléments transposables | Arabidopsis thaliana | Homo sapiens | Saccharomyces cerevisiae |
| nombre de copies classe I -LTR | 1594 | 443.000 | 331 |
| nombre de copies classe I -non LTR | 515 | 2.426.000 | 0 |
| nombre de copies classe II | 2203 | 294.000 | 0 |
Les valeurs ci-dessus ne sont qu'indicatives. Elles évoluent en effet au fur et à mesure que les génomes sont re-séquençés, analysés structuralement et enfin ré-annotés. Base de données "ACLAME" : "A CLAssification of geneticMobile Elements" |
|||
| g. Famille Alu
Famille de séquences répétées issue d'un processus de rétrotransposition. Les séquences Alu contiennent un site de reconnaissance pour l'enzyme de restriction Alu I.
La famille Alu est un exemple de SINE ("Short Interspersed Nuclear Elements" - voir ci-dessus). La plupart sont des rétroséquences issues d'une réverse transcription de l'ARN. |
|
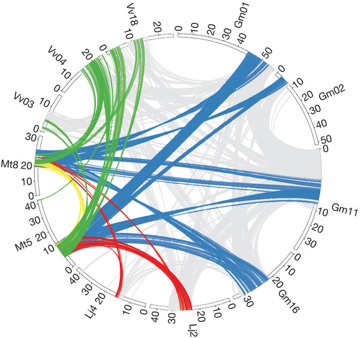
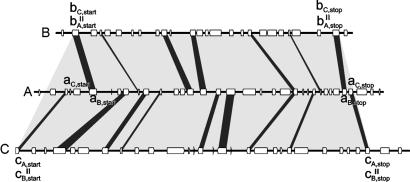
h. La synténie Localement l'ordre des gènes sur un même chromosome tend à être conservé sur des millions d'années : c'est le phénomène de synténie. La synténie est donc la présence simultanée sur le même chromosome de deux ou plusieurs loci. La synténie est utilisée en génomique comparative (cartes physiques comparatives) pour décrire la conservation de l'ordre des gènes entre deux espèces apparentées. Les comparaisons entre espèces éloignées phylogénétiquement éloignées révèlent une perte de synténie. Figure ci-dessous : macrosynténie entre les génomes de Medicago truncatula et L. japonicus et Glycine max.
Source : Young et al. (2011) |
|
i. Gène candidat C'est un gène dont on suppose l'implication dans un effet biologique. C'est donc un gène qui gouverne une part importante de la variabilité d'un caractère. Plusieurs approches (qui peuvent être combinées) existent pour identifier un gène candidat :
|
|
j. Les microsatellites Une séquence d'ADN dite microsatellite (ou "simple sequence repeats" - SSR, "short tandem repeats" - STR) est formée par une répétition continue de motifs de 2 à 10 nucléotides (exemple : le motif CAGT). Ils sont très abondants chez les eucaryotes : un microsatellite peut être présent à des milliers d'exemplaires dans le génome. Chez les végétaux supérieurs, il y aurait en moyenne un microsatellite tous les 50 kb. Les microsatellites sont présents sur l'ensemble du génome, le plus fréquemment au niveau des introns et des exons des gènes. La localisation des microsatellites sur le génome est relativement conservée entre des espèces phylogénétiquement proches. Le polymorphisme des microsatellites peut être utilisé comme marqueur génétique afin d'identifier un individu. Les régions flanquantes des microsatellites servent d'amorces pour la réaction de polymérisation en chaîne. En effet, si un microsatellite donnné n'est pas spécifique d'un locus, en revanche ses régions flanquantes le sont. Une paire d'amorces spécifique de ces régions flanquantes permettra donc l'amplification spécifique de ce seul microsatellite. |
|
k. Marqueur génétique C'est une séquence d'ADN polymorphe utilisée pour baliser le génome et obtenir une carte génétique. Les nouvelles biotechnologies permettent l'analyse directe du polymorphisme des séquences d'ADN. Il existe différentes sortes de marqueurs :
|
|
2. Pourquoi Arabidopsis thaliana comme plante modèle ? Taxonomie : Eukaryota; Viridiplantae; Streptophyta; Embryophyta; Tracheophyta; Spermatophyta; Magnoliophyta; eudicotyledons; core eudicotyledons; rosids; eurosids II; Brassicales; Brassicaceae; Arabidopsis; Arabidopsis thaliana L'espèce Arabidopsis thaliana est de la famille du chou (Brassicaceae). Elle a été découverte au 16è siècle par Johannes Thal. Elle est aussi nommée arabette rameuse (ou "Wall cress", "mouse-ear cress", "thale-cress" en anglais). Plus de 750 accessions naturelles d'Arabidopsis thaliana ont été collectées dans le monde et sont disponibles dans les 2 centres de semences : ABRC et le NASC ("Nottingham Arabidopsis Stock Centre"). Quelques caractéristiques:
Source : TAIR |
|
La complexité et la grande taille des génomes des plantes cultivées rendent difficiles leur analyse génétique et moléculaire. Mais comme il y a des similitudes entre les génomes des différentes espèces (cultivées ou non), on a choisi une plante ayant le plus petit génome possible, pour l'analyser et utiliser les informations obtenues pour l'étude des génomes plus complexes et plus grands. Le choix de la communauté scientifique internationale s'est porté sur Arabidopsis thaliana qui présente de nombreux avantages :
En conséquence, Arabidopsis thaliana est une espèce modèle pour les études de génétique et de génomique fonctionnelle. La séquence complète du génome de l'accession Columbia a été publiée en 2000 ("The Arabidopsis Genome Initiative" Nature 408, 796 - 815). Le décryptage du génome d'une plante modèle comme Arabidopsis thaliana est capital du fait de la ressemblance entre le génome de cette espèce modèle et celui d'espèces cultivées apparentées. En effet, les gènes sont souvent organisés de la même manière sur les chromosomes, et placés dans le même ordre (synténie). Par ailleurs, l'absence d'intérêt économique particulier d'Arabidopsis thaliana est un point favorable pour l'échange de données entre équipes de recherche. Les informations issues d'un génome modèle sont donc une une base solide pour analyser les chromosomes d'espèces cultivées dont les génomes sont plus complexes et plus grands . Quelques exemples des retombées chez les espèces cultivées de l'identification de la fonction des gènes chez Arabidopsis thaliana:
|
|
3. Le génome d'Arabidopsis thaliana Les cartes physiques des 5 chromosomes ont été complètement déterminées en 1997. Le génome d'Arabidopsis thaliana a été séquencé par une méthode ordonnée ("top-down") et publié en 2000 : Arabidopsis Genome Initiative (2000) "Analysis of the genome sequence of the flowering plant Arabidopsis thaliana". Nature 408, 796 - 815. Le génome d'Arabidopsis thaliana sert de modèle pour l'un des deux grand groupes de plantes à fleurs, les dicotylédones.
|
| chromosome | longueur (Mpb) | teneur en [G+C] (%) | Protéines codées |
| 1 (métacentrique) | 30,0 | 35,7 | 8521 |
| 2 (acrocentrique) | 20,0 | 35,8 | 5182 |
| 3 (submétacentrique) | 23,0 | 36,3 | 6432 |
| 4 (acrocentrique) | 19,0 | 36,2 | 4934 |
| 5 (submétacentrique) | 27,0 | 35,9 | 7545 |
| chloroplastique | 0,15 | 36,3 | 85 |
| mitochondrial | 0,37 | 44,8 | 117 |
|
Les valeurs ci-dessus ne sont qu'indicatives. Elles évoluent en effet au fur et à mesure que les génomes sont re-séquençés, analysés structuralement et enfin ré-annotés. |
|||
Remarque : ces chiffres ne sont qu'indicatifs car ils évoluent au fur et à mesure que l'ensemble du génome est ré-analysé et donc ré-annoté. |
Le degré de redondance (la duplication de gènes ou de régions identifiés sur un chromosome) est élevé : 24 régions d'une taille supérieure à 100 kb sont dupliquées, soit 65,6 Mb, soit 58% du génome (figure ci-contre).
Source : "Précis de génomique" - Gibson & Muse (2004) - Ed. De Boeck Université Celà suggère qu'Arabidopsis thaliana soit passé récemment par un état tétraploide avec ensuite, une perte d'une partie des gènes dupliqués. 3 duplications :
Source : Simillion et al. (2002) Par ailleurs, un transfert du génome mitochondrial vers le noyau a certainement eut lieu récemment chez Arabidopsis thaliana puisqu'une région de 620 kb identique à 99% au génome mitochondrial a été identifiée sur le chromosome 2. |
| Exemples de génomes séquencés ou en cours de séquençage - Aller à "GOLD : Genomes OnLine Database". | |
| Génomes de plantes séquencés | Arabidopsis thaliana / Riz (Oryza sativa) / Peuplier (Populus trichocarpa) / Vigne (Vitis vinifera) / Soja (Glycine max.) / Blé (Triticum aestivum) |
| Génomes séquencés ou séquençages bien avancés (streptophytes) | Maïs (Zea mays) / Luzerne (Medicago truncatula) / Orge (Hordeum vulgare) / Tomate (Solanum lycopersicum) / Choux (Brassica napus) / Piment (Capsicum annuum) / Tabac (Nicotiana tabacum) / Café (Coffea arabica) / Pin (Pinus radiata) / Lotus japonica / Eucalyptus / Sorghum bicolor / Physcomitrella patens(mousse) ... |
| Génomes séquencés ou séquençages bien avancés (chlorophytes) |
Chlamydomonas reinhardtii (algue) / Micromonas pusilla (algue) / Ostreococcus lucimarinus (algue) / Ostreococcus tauri (algue) / Botryococcus braunii/ Chlorella vulagaris / Dunaliella salina / Nephroselmis olivacea / Prototheca wickerhamii ... |
|
Exemples de l'intéret des génomes complétement séquencés et clairement annotés Voir un cours sur l'annotation. a. En premier lieu, celà permet de les comparer à d'autres génomes moins bien (ou pas du tout) séquencés et/ou annotés. b. Les duplications doivent être prises en compte si l'on veut créer de nouveaux traits agronomiques par la modification de l'expression d'un ou de plusieurs gènes, car ceux-ci peuvent posséder des copies ailleurs dans le génome. Les séquences répétées constituent un élément dynamique du génome qui est impliqué dans la génération d'une part importante de la variation allélique exploitée par les sélectionneurs. c. Si la séquence du génome est "ancrée" par rapport aux cartes physique et génétique, pour un trait agronomique cartographié dans un intervalle génétique, il est possible de rechercher les séquences génomiques correspondantes, et l'ensemble des gènes annotés dans l'intervalle. d. Les informations disponibles sur la fonction de ces gènes permettent alors de retenir un ou plusieurs gènes candidats pour le trait considéré. La validation d'un candidat peut notamment passer par l'étude de son expression et de sa variabilité allélique dans plusieurs variétés différant pour le trait étudié, ainsi que par l'étude de plantes mutées ou génétiquement transformées dans lesquelles l'expression du gène est éteinte ou augmentée. e. Les séquences des génomes révélent une grande quantité de nouveaux marqueurs moléculaires de type microsatellite qui permettent d'affiner l'analyse génétique. Ces nouveaux marqueurs facilitent et accélérent la création de nouvelles variétés par sélection assistée par marqueurs : grâce à des marqueurs plus étroitement associés au locus responsable d'un caractère, on suit mieux sa transmission au fil des croisements et on réduit le matériel génétique indésirable transmis en même temps que lui. Lorsque le caractère est associé à un ou plusieurs allèle(s) identifié(s), la séquence de cet (ces) allèle(s) fournit un marqueur absolu. f. On peut identifier des polymorphismes de type SNP sans risque de confusion avec des erreurs de séquençage. |
|
Les génomes mitochondriaux des plantes Voir un cours sur la mitochondrie. Ils se distinguent de ceux des autres eucaryotes. Leur taille est plus importante et beaucoup plus variable (de 200 kb chez les brassicacées à 2500 kb chez les cucurbitacées, contre 16 à 20 kb chez les mammifères). La caractéristique de ces génomes est la présence de séquences répétées (impliquées dans des recombinaisons entre molécules d'ADN mitochondrial) variables en taille, en nombre de répétition et en orientation. Cependant, malgré leur grande variabilité en taille, les génomes mitochondriaux des plantes semblent avoir le même contenu en information génétique : de 100 à 120 gènes (inclus ceux qui codent pour les ARN de transfert et les ARN ribosomiques). Il existe un processus propre aux mitochondries végétales : l'édition des messagers. Le transcrit primaire peut subir des modifications post-transcriptionnelles qui changent spécifiquement certaines cytidines en uraciles. Les codons de l'ARN messager sont donc modifiés. |
|
Le génome chloroplastique Voir un cours sur le chloroplaste. Les chloroplastes possèdent plusieurs copies d'un génome circulaire mesurant de 120 à 217 kb (150 kb en moyenne) chez les plantes supérieures. Le séquençage complet de l'ADN chloroplastique de plusieurs plantes très éloignées au sens évolutif (algues, bryophytes, gymnospermes, angiospermes) a révélé que chez les plantes terrestres, l'organisation de ce génome circulaire est remarquablement conservée. Hormis pour certaines légumineuses et des conifères, le génome chloroplastique est caractérisé par la duplication d'une région contenant l'ADN ribosomique chloroplastique. Les gènes de l'ADN chloroplastique se répartissent en deux catégories :
L'ADN chloroplastique a un taux d'évolution assez lent. Ainsi, le séquençage de parties de l'ADN chloroplastique (comme le gène rbcL) permet de retracer l'évolution des grandes familles de plantes via des reconstructions phylogénétiques. |
|
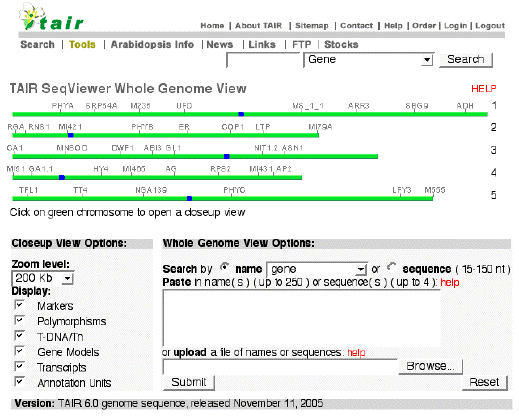
4. Bases de données relatives à Arabidopsis thaliana a. TAIR : The Arabidopsis Information Resource Figure ci-dessous : page d'accueil de "Sequence Viewer", logiciel pour la recherche et la visualisation de séquences de gènes, de séquences transcrites, de marqueurs (et bien d'autres informations) sur les 5 chromosomes.
|
|
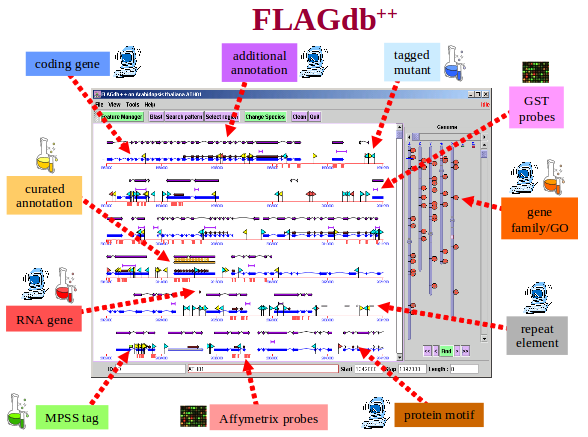
Le but de la base de données Flagdb++ est de développer une interface de travail basée sur l'intégration de données autour du génome d'Arabidopsis thaliana, afin d'aider les utilisateurs à comprendre la fonction biologique des gènes végétaux en les considérant dans un contexte large : une famille multigénique et/ou un réseau fonctionnel.
FLAGdb++ est développée de façon générique pour pouvoir s'appliquer à différents génomes et pour pouvoir stocker, organiser, visualiser et exploiter de nombreux types de données de génomique. L'intégration des données se fait par leur association aux séquences nucléiques, indépendamment les unes des autres. Elle concerne actuellement :
|
|
c. AraCyc (Arabidopsis thaliana Biochemical Pathways) Exemple de recherche de données sur le cycle de Calvin et la RuBisCO. Aller à AraCyc. Taper "Calvin" dans la fenêtre "Search" en haut à droite. Activer le lien vers les gènes codant la RuBisCO en passant la souris sur la flèche entre "D-ribulose 1,5-bisphosphate" et "3-phosphoglycérate". |
|
d. Le centre de ressources Arabidopsis thaliana pour la génomique (INRA Versailles) Ce centre de ressources propose une collection de plusieurs dizaines de milliers de mutants d'insertion indépendants (mutants pour environ 80% des gènes) obtenus par mutagenèse T-DNA (insertion d'une cassette d'ADN dans l'écotype WS).
|
|
5. Données sur le riz (Oryza sativa) - Principale source : Genoscope Intérêts du séquençage d'une céréale Voir un cours sur le séquençage. Les céréales (riz, blé, orge, maïs, sorgho, millet, canne à sucre,... ) constituent une part essentielle de l'alimentation humaine. Cependant, les progrès en agronomie que requièrent l'explosion démographique mondiale, le recul des terres arables et le changement climatique, nécessitent une nouvelle approche : le séquençage du génome d'une céréale. En effet, à partir de la séquence génomique, on peut :
|
a. Intérêt du génome du riz par rapport à celui des autres céréales Le riz possède le plus petit génome parmi toutes les céréales (430 millions de nucléotides). Le génome du maïs est 5 fois plus gros, celui du blé est 40 fois plus gros. D'une céréale à une autre, de grands blocs de gènes homologues sont retrouvés et leur arrangement est relativement conservé (synténie). Le riz est donc un bon choix pour caractériser les gènes des autres céréales et les associer à tel ou tel trait agronomique. Le riz sert de génome modèle pour l'un des deux grand groupes de plantes à fleurs, les monocotylédones (Arabidopsis thaliana servant de génome modèle pour l'autre groupe, les dicotylédones). Le choix du riz s'appuie aussi sur des ressources pour l'approche génomique: on dispose d'excellentes cartes génétiques et des techniques de transformation génétique qui font du riz la céréale la plus facile à transformer. Il existe un très grand nombre de variétés de riz : plus de 90 000 variétés traditionnelles et espèces sauvages de riz sont gérées par l'IRRI ("International Rice Research Institute"). Ces variétés sont adaptées à des conditions environnementales très diverses (des sols secs en régions tempérées aux cultures inondées en régions tropicales). Ainsi, le riz est déja un modèle dans plusieurs autres domaines:
|
b. Intérêt du génome du riz en lui-même Le riz constitue la base quotidienne de l'alimentation de plus de la moitié de l'humanité. La production de riz représente 30% de la production mondiale de céréales. Elle a doublé en trente ans, notamment avec l'introduction de nouvelles variétés. Cependant cette croissance de production suit difficilement celle de la consommation : 4,6 milliards de personnes dépendront du riz pour leur consommation quotidienne en 2025 (contre 3 milliards actuellement). En parallèle, les petits producteurs disposent de terrains de culture moins favorables (par exemple saumâtres) et l'approvisionnement en eau est un problème de plus en plus aigu. Ainsi, la connaissance du génome du riz est extrêmement précieuse pour les sélectionneurs dans le but d'augmenter le rendement et créer de nouvelles variétés résistantes aux maladies, aux ravageurs, à la sécheresse ou à la salinité. Cette connaissance permet de repérer les gènes intéressants du point de vue agronomique et permet de rechercher les variants alléliques avantageux dans la collection de l'IRRI. Cela devrait aussi permettre de répondre aux inquiétudes liées à l'érosion génétique du fait de l'abandon des variétés traditionnelles au profit des variétés à haut rendement. La génomique du riz pourrait déboucher sur le transfert rapide de traits avantageux à des variétés localement adaptées. |
c. Littérature et base de données sur le génome du riz Le génome complet du riz a été publié en 2002 :
Base de données pour l'annotation du génome du riz : RAPDB ("Rice Annotation Project Database"). |
d. Bénéfices du séquençage complet du génome du riz La prédiction des gènes dépend de la qualité de la séquence car on ne peut tous les identifier à partir d'une ébauche. Seule une vision exhaustive du bagage de gènes du riz permet de déterminer si une voie métabolique est absente, ce qui est capital dans la perspective de l'ingénierie métabolique d'un organisme en général, du riz en particulier. L'analyse des duplications et des séquences répétées dépend aussi complètement de la qualité de la séquence :
A ces bénéfices s'ajoute ceux d'une séquence "ancrée" par rapport aux cartes physique et génétique : pour un trait agronomique cartographié dans un intervalle génétique, il est dés lors possible de rechercher les séquences génomiques correspondantes et les gènes qui sont annotés dans cet intervalle. Cela permet alors de retenir un ou plusieurs gènes dits candidats pour le trait considéré. La validation d'un gène candidat passer entre autre :
Enfin, la séquence du génome du riz :
Ces nouveaux marqueurs facilitent la création de nouvelles variétés par sélection assistée par marqueurs. En effet, des marqueurs étroitement associés au locus responsable d'un caractère permettent de suivre sa transmission au fil des croisements et ainsi de réduire le matériel génétique indésirable transmis en même temps que lui. |
|
6. Medicago truncatula et le soja a. Medicago truncatula Le génome de Medicago truncatula a été séquencé en 2011 (Nevin et al. "The Medicago Genome Provides Insight into the Evolution of Rhizobial Symbioses" Nature). Medicago truncatula est une plante diploïde vraie, autogame avec un petit génome qui comporte 8 chromosomes. Medicago truncatula fait partie de la famille des Fabaceae (anciennement légumineuse). Les légumineuses jouent un rôle économique majeur : elles constituent une source importante de protéines végétales pour l’alimentation animale et humaine : son rôle crucial dans la fixation de l'azote dans les sols constitue une part importante de l'alimentation fourragère. La culture des légumineuses ne nécessite pas de fertilisation azotée ce qui représente un avantage économique mais aussi environnemental. La capacité des légumineuses à fixer l’azote atmosphérique est un caractère unique parmi les plantes cultivées. Elle résulte de la symbiose avec des bactéries du sol appelées Rhizobium qui induisent la formation des structures appelées nodosités au niveau des racines de la plante. Ces bactéries produisent une enzyme, la nitrogénase, dont les plantes sont dépourvues et qui permet aux légumineuses de fixer l’azote atmosphérique. En échange, la plante fournit les éléments nutritifs nécessaires au développement des bactéries. Le séquençage du génome de Medicago truncatula a révélé une duplication de l'ensemble du génome au moment de l'apparition des légumineuses, il y a environ 60 millions d'années. Cette duplication a joué un rôle majeur dans la formation du génome de Medicago truncatula en favorisant la mise en place d’un programme génétique permettant une vie symbiotique avec Rhizobium. En effet, grâce à cette duplication du génome, des gènes impliqués dans une symbiose beaucoup plus ancienne avec des champignons mycorhiziens ont évolué et donné naissance à des gènes impliqués dans la symbiose fixatrice d'azote. Medicago truncatula est donc, du point de vue de la recherche, la légumineuse modèle (proche de la luzerne cultivée) pour l'étude la génétique moléculaire de la symbiose fixatrice d'azote avec la bactérie du sol Rhizobium et la symbiose endomycorhizienne. Au cours de son cycle de développement, la plante passe successivement d'un métabolisme assimilateur de l'azote, à un métabolisme mobilisateur assurant le recyclage de l'azote protéique stocké dans l'ensemble de ses organes. De cette manière, la plante assure d'abord sa croissance puis gère ensuite ses réserves azotées et carbonées accumulées pour assurer la formation des organes de réserve. La remobilisation de l'azote et du carbone pendant la sénescence, conditionne le rendement en biomasse ainsi que les teneurs en protéines et en glucides des organes récoltables. Medicago truncatula est très proche, d'un point de vue phylogénétique, de la plupart des légumineuses cultivées en Europe comme le pois protéagineux, la féverole, la luzerne ou les trèfles. Il existe une forte conservation de l'ordre dans lequel les gènes sont situés sur les chromosomes de ces espèces (conservation synténique). Le séquençage du génome de Medicago truncatula permet de déterminer l'ordre de la majorité des gènes sur les 8 chromosomes. Celà devrait grandement faciliter la localisation des gènes importants chez les légumineuses cultivées. L'amélioration génétique des légumineuses est indispensable pour permettre leur introduction plus fréquente dans les rotations de cultures pour développer des systèmes durables et moins consommateurs d’intrants, en particulier les nitrates, dont la production est coûteuse en énergie. |
b. Le soja Le génome du soja (légumineuse - 1,1 milliard de paires de base d'ADN) a été séquencé (Schmutz et al., 2010 - méthode "shotgun") en 2010 et on a mis en évidence 90 caractères distincts affectant le développement de la plante, la productivité, la résistance aux maladies, la qualité des semences et la nutrition. En plus de la cartographie du génome (plus de 46.000 gènes dont 34,000 sont orthologues - 73% de copies / la plus grande partie du génome du soja est composée d'éléments transposables), une base de données de facteurs de transcription régulant l'expression des gènes a été créée (SoybeanDB). Avec un poids économique de près de 30 milliards de dollars aux Etats-Unis, le soja est la deuxième culture la plus rentable après le maïs. |
| c. Plantes et algues dont le génome a été séquencé (Novembre 2011) | ||
|
|
Aller à GOLD. |
| 7. Liens Internet et références bibliographiques | |
|
Arabidopsis Genome Initiative (2000) "Analysis of the genome sequence of the flowering plant Arabidopsis thaliana". Nature 408, 796 - 815 Simillion et al. (2002) "The hidden duplication past of Arabidopsis thaliana" P.N.A.S. 99, 13627 - 13632 Nevin et al. (2011) "The Medicago Genome Provides Insight into the Evolution of Rhizobial Symbioses" Nature 480, 520 - 524 |
|
|
MASC Annual Reports : Annual reports summarize yearly progress of the global Arabidopsis research community and include reports of the member countries and MASC subcommittees as well as recommendations to the community by the MASC for the upcoming year. "Génomique structurale, génomique fonctionnelle" - Bernard Dujon - Conférence "Université de tous les savoirs" (Vidéo et documents à télécharger) |
|
|
The Arabidopsis Information Resource (TAIR) Arabidopsis Biological Resource Center (ABRC) European Arabidopsis Stock Centre (NASC) Riken BioResource Center Arabidopsis thaliana Resource Centre for Genomics 1001Genomes Project (Whole-genome sequence variation in 1001 accessions of A. thaliana) |
|
|
FLAGdb++ : base de données intégrative en génomique végétale (Génoplante - INRA - CNRS) Samson et al. (2004) "FLAGdb++: a database for the functional analysis of the Arabidopsis genome" Nucleic Acids Res. 32 - Database issue: D347 - 350 |
|
|
AraCyc : Arabidopsis thaliana Biochemical Pathways Zhang et al. (2005) "MetaCyc and AraCyc. Metabolic pathway databases for plant resaerch" Plant Physiol. 138, 27 - 3 MetaCyc : database of nonredundant, experimentally elucidated metabolic pathways from more than 300 different organisms. MetaCyc is curated from the scientific experimental literature Krieger et al. (2004) "MetaCyc: A multiorganism database of metabolic pathways and enzymes" Nucleic Acids Res. 32, D438 - D442 Horan et al. (2005) "Genome cluster database. A sequence family analysis platform for Arabidopsis and rice" Plant Physiol. 138, 47 - 54 Bolle et al. (2011) "Perspectives on Systematic Analyses of Gene Function in Arabidopsis thaliana: New Tools, Topics and Trends" Curr. Genomics 12, 1 - 14 |
|
|
Le génome du riz et du soja
Yu et al. (2002) "A Draft Sequence of the Rice Genome (Oryza sativa L. ssp. indica)" Science 296, 79 Goff et al. (2002) "A Draft Sequence of the Rice Genome (Oryza sativa L. ssp. japonica)" Science 296, 92 International Rice Genome Sequencing Project (2005) "The map-based sequence of the rice genome" Nature 436, 793 - 800 Schmutz et al. (2010) "Genome sequence of the palaeopolyploid soybean" Nature 463, 178-183 |
|
![]()


{kind=link}