| Les structures secondaires des protéines |
| Tweet |
|
|
1. Introduction 2. Les hélices
3. Les feuillets
|
4. Les structures secondaires mixtes
5. Tours et boucles 6. Les méthodes de prédiction de structures secondaires 7. Liens Internet et références bibliographiques |
|
1. Introduction En 1953, la structure en double hélice de l'ADN a été résolue par F. Crick , J. Watson et M. Wilkins (Prix Nobel 1962). L'information dans l'ADN est sous forme linéaire. En conséquence, les molécules d'ADN ont une structure semblable. En revanche, chacune des milliers de protéines qui existent dans la cellule doit reconnaitre spécifiquement un (ou quelques) ligand(s). Cette reconnaissance s'effectue grâce à de subtiles interactions entre la structure tridimensionnelle de la protéine et celle(s) du (des) ligand(s). Une telle diversité d'interactions aussi précises ne peut être obtenue qu'avec des structures de protéines extrêmement variées et irrégulières. Au sein de la structure tridimensionnelle des protéines, il existe cependant des "sous"-structures aux caractéristiques régulières : les structures secondaires. Voir les définitions de toutes les structures secondaires recencées : base de données CATH (glossaire) |
|
Visualisation du répresseur ROP de Escherichia coli à une résolution de 1,7 Å Code PDB : 1ROP
|
|
Hélice 310 :
Hélice π :
|
| Hélices polyproline I et II | |
| Poly - Pro I | Poly - Pro II |
| liaisons peptidiques cis | liaisons peptidiques trans |
| hélice droite avec 3.3 résidus par tour | hélice gauche avec 3 résidus par tour |
| Φ = - 83° | Φ = - 78° |
| Script Python : diagramme de Ramachandran d'une séquence polypeptidique | |
|
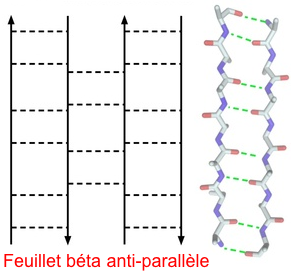
3a. Structures β antiparallèles Les structures β antiparallèles forment le 2ème grand groupe de domaines de protéines.
Source : Expasy - les liaisons hydrogène sont figurées par les pointillés C'est aussi le plus diversifié puisqu'il comprend des enzymes, des protéines de transport, des anticorps, des protéines d'enveloppe de virus ...).
Les feuillets β ont une torsion classique et quand 2 de ces feuillets β sont assemblés, ils forment une structure en tonneau ("β barrel"). Voir l'exemple des porines. |
|
Visualisation de la superoxide dismutase de Bos taurus à une résolution de 1,85 Å Code PDB : 1E9O |
| type de structure | valeurs des angles en degrés |
nombre moyen de résidus par tour | translation par résidu (angstroem) | ||
| Φ | Ψ | ω | |||
| hélice α | - 57 | - 47 | 180 | 3,6 | 1,5 |
| hélice 310 | - 49 | - 26 | 180 | 3,0 | 2,0 |
| hélice π | - 57 | - 70 | 180 | 4,4 | 1,15 |
| hélice polyproline I | - 83 | + 158 | 0 | 3,38 | 1,9 |
| hélice polyproline II | - 78 | + 149 | 180 | 3,0 | 3,12 |
| brin β parallèle | - 119 | + 113 | 180 | 2,0 | 3,2 |
| brin β antiparallèle | - 139 | + 135 | - 178 | 2,0 | 3,4 |
| Source : base de données PROWL --> consulter l'item : "Residue hydrogen bonding" | |||||
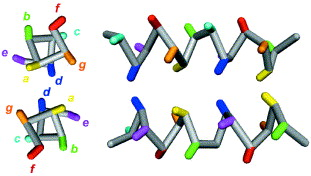
Structure en spirale d'hélices α "coiled-coil" Le motif structural α-hélicoïdal multi-spiralé appelé "coiled-coil" permet l'oligomérisation des sous-unités d'un grand nombre de protéines. Les prédictions basées sur l'analyse des séquences primaires indiquent que 2 à 3 % des acides aminés participent à ce type de structure. Ce motif structural se compose de deux à cinq hélices α parallèles amphiphiles (présence d'acides aminés hydrophiles et d'acides aminés hydrophobes / apolaires) qui s'enroulent les unes autour des autres pour former un super-enroulement. Exemples : vimentine, protéines du complexe SNARE Les séquences en acides aminés des protéines qui adoptent un super-enroulement de pas gauche sont caractérisées par une périodicité de 7 résidus (répétition heptade) avec des résidus apolaires préférentiellement en première (a - figure ci-dessous) et quatrième position (d).
Source : Burkhard et al. (2001) Les séquences en acides aminés des protéines qui adoptent un super-enroulement de pas droit sont caractérisées par une périodicité de 11 résidus (répétition undecade). La stabilité de ces bobines super-enroulées est principalement la conséquence d'un empaquetage des chaînes latérales des acides aminés apolaires dans un coeur hydrophobe. |
|
4. Les structures secondaires mixtes 4a. Les structures alpha/beta La structure de domaines de protéines la plus fréquente et la plus régulière est celle du domaine alpha/béta (α/β) constitué d'un feuilletβ central parallèle ou mixte entouré d'hélices α. Toutes les enzymes de la glycolyse ont ce type de structure. Dans ces domaines, les sites de liaison des ligands sont formés par des boucles de jonction. Celles-ci ne contribuent pas à la stabilité de la structure mais participent à la fléxibilité conformationnelle nécessaire pour les mouvements liés à l'activité catalytique (à la fixation des ligands de manière générale). La glycéraldehyde 3-phosphate déshydrogénase est une enzyme de la glycolyse. Elle catalyse la seule réaction d'oxydo-réduction de cette voie métabolique. Elle utilise la nicotinamide adénine dinucléotide (NAD+). |
|
Visualisation de la glyceraldehyde 3-phosphate déshydrogénase de Thermus Thermophilusà une résolution de 2,6 Å Code PDB : 1VC2 |
|
Les tours C'est la structure qui connecte deux brins β antiparallèles. Les tours sont généralement courts : 2 à 4 acides aminés en dehors des brins. Les conformations possibles ont été classées selon les valeurs des angles Φ et Ψ des différents acides aminés. Le tableau ci-dessous donne les types les plus importants, 1 ou 2 acides aminés dans le tour. Le dernier acide aminé du premier brin est appelé i et le premier acide aminé du second brin est appelé i+2. |
| Type de tours | sous - type | Φi+1 | Ψi+1 | Φi+2 | Ψi+2 |
| γ | classique | + 70 à + 85 | - 60 à - 70 | ||
| inverse | + 70 à + 85 | - 60 à - 70 | |||
| β | I | - 60 | - 30 | - 90 | 0 |
| I' | +60 | +30 | + 90 | 0 | |
| II | - 60 | + 120 | + 80 | 0 | |
| II' | + 60 | - 120 | - 80 | 0 | |
| D'autres sous-types (de II, II' jusqu'à VIII) ont été recensés. | |||||
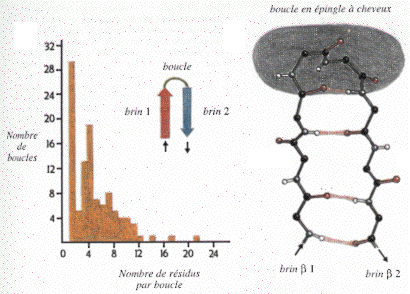
Les boucles Les structures des protéines sont souvent des combinaisons d'hélice et de feuillets reliées par des boucles de longueurs trés variables : de 1 à 12 résidus (voire jusqu'à 22) avec le plus fréquemment 1, 3, 4 ou 7 résidus. La comparaison de structures tridimensionnelles montre que les boucles adoptent un nombre limité de conformations.
Source : "Introduction à la structure des protéines" (1996) Branden & Tooze - Ed. DeBoeck Université Elles participent donc à la formation :
Les boucles sont généralement à la surface des protéines. Les groupements C=O et NH (liaison peptidique) ne forment en général pas de liaisons hydrogène entre eux mais plutôt avec les molécules du solvant (l'eau). Les régions des boucles exposées au solvant sont riches en acides aminés hydrophiles, chargés ou polaires. Ce sont des régions privilégiées d'insertions ou de délétions entre séquences homologues. Par ailleurs, beaucoup d'introns au niveau des gènes correspondent à des boucles. Les boucles qui relient 2 brins β antiparallèles sont dites en "épingle à cheveux". Voir un exemple de structure hélice - boucle - hélice : le motif "EF-hand" de la calmoduline. |
|
6. Les méthodes de prédiction de structures secondaires La première d'entre elles, la méthode de prédiction de Chou et Fasman (1974-1978) est une méthode statistique. Des tables d'occurence des acides aminés dans les différents états structuraux ont été établies. Chou et Fasman ont observé les 29 structures 3D connues à cette époque (on en dénombre environ 120.000 en 2016) et ils ont affecté chaque acide aminé à un état structural : hélice, brin, apériodique ou coude. |
| Nom | Nombre | hélice | feuillet | apériodique | coude |
| Arg | 142 | 53 | 26 | 63 | 40 |
| Cys | 94 | 25 | 22 | 47 | 33 |
| Gln | 162 | 68 | 35 | 59 | 47 |
| Tyr | 184 | 48 | 53 | 83 | 62 |
| Val | 357 | 144 | 119 | 94 | 53 |
| Total | 4741 | 1798 | 930 | 2013 | 1400 |
| Source : G. Deléage | |||||
|
Ci-dessous : table de Chou et Fasman - Source : algorithme décrit à la base de données PROWL Nom P(α) P(β) P(turn) Alanine 142 83 66 Arginine 98 93 95 Aspartic Acid 101 54 146 Asparagine 67 89 156 Cysteine 70 119 119 Glutamic Acid 151 037 74 Glutamine 111 110 98 Glycine 57 75 156 Histidine 100 87 95 Isoleucine 108 160 47 Leucine 121 130 59 Lysine 114 74 101 Methionine 145 105 60 Phenylalanine 113 138 60 Proline 57 55 152 Serine 77 75 143 Threonine 83 119 96 Tryptophan 108 137 96 Tyrosine 69 147 114 Valine 106 170 50 Sur la base de cette table, Chou et Fasman ont établit la table des paramètres conformationnels Pα, Pβ et P(coude) en calculant les rapports des fréquences. Exemple pour Tyr : Pβ (Tyr) = [(53 / 184) / (930 / 4741)] = 147 Il existe maintenant un trés grand nombre de méthodes de prédiction dont la plupart sont répertoriés à EXPASY ("ExPASy Proteomics tools"). A partir des outils du site EXPASY, s'exercer à la prédiction de structures secondaires avec la séquence de la protéine LEA ("Late Embryogenesis Abundant") de Arabidopsis thaliana: >1YYCA ghhhhhhleasadekvveekasvisslldkakgffaeklaniptpeatvddvdfkgvtrdgvdyhakvsv knpysqsipicqisyilksatrtiasgtipdpgslvgsgttvldvpvkvaysiavslmkdmctdwdidyq ldigltfdipvvgditipvstqgeiklpslrdff Exemple de logiciel "GOR" (Garnier et al, 1996) - faire un copier / coller de la séquence sans le n° d'accession. Voir la structure 3D de cette protéine LEA : PDB 1YYC. |
Il existe des "calculateurs" de propriétés physico-chimiques des protéines. Ils s'appuient sur des masses de données colossales donc ont une bonne signification statistique. Exemple Composition Profiler : "A web-based tool that automates detection of enrichment or depletion patterns of individual amino acids or groups of amino acids classified by several physico-chemical and structural properties such as aromaticity, charge, polarity, hydrophobicity, flexibility, surface exposure, solvation potential, interface propensity, normalized frequency of occurrence in helices, structures, and coils, linker and disorder propensities, size and bulkiness." |
Residue SwissProt PDB S25 Surface DisProt Ala (A) 7.89 ± 0.05 7.70 ± 0.08 6.03 ± 0.13 8.10 ± 0.35 Arg (R) 5.40 ± 0.04 4.93 ± 0.06 6.56 ± 0.13 4.82 ± 0.23 Asn (N) 4.13 ± 0.04 4.58 ± 0.06 6.23 ± 0.15 3.82 ± 0.27 Asp (D) 5.35 ± 0.03 5.83 ± 0.05 8.18 ± 0.10 5.80 ± 0.30 Cys (C) 1.50 ± 0.02 1.74 ± 0.05 0.78 ± 0.04 0.80 ± 0.08 Gln (Q) 3.95 ± 0.03 3.95 ± 0.05 5.21 ± 0.09 5.27 ± 0.37 Glu (E) 6.67 ± 0.04 6.65 ± 0.07 8.70 ± 0.17 9.89 ± 0.61 Gly (G) 6.96 ± 0.04 7.16 ± 0.07 7.06 ± 0.11 7.41 ± 0.40 His (H) 2.29 ± 0.02 2.41 ± 0.04 2.60 ± 0.06 1.93 ± 0.11 Ile (I) 5.90 ± 0.04 5.61 ± 0.06 2.77 ± 0.07 3.24 ± 0.13 Leu (L) 9.65 ± 0.04 8.68 ± 0.08 5.11 ± 0.08 6.22 ± 0.25 Lys (K) 5.92 ± 0.05 6.37 ± 0.08 9.75 ± 0.16 7.85 ± 0.45 Met (M) 2.38 ± 0.02 2.22 ± 0.04 1.13 ± 0.04 1.87 ± 0.10 Phe (F) 3.96 ± 0.03 3.98 ± 0.04 2.38 ± 0.05 2.44 ± 0.13 Pro (P) 4.83 ± 0.03 4.57 ± 0.05 5.63 ± 0.10 8.11 ± 0.63 Ser (S) 6.83 ± 0.04 6.19 ± 0.06 6.87 ± 0.13 8.65 ± 0.43 Thr (T) 5.41 ± 0.02 5.63 ± 0.05 6.08 ± 0.11 5.56 ± 0.24 Trp (W) 1.13 ± 0.01 1.44 ± 0.03 1.33 ± 0.05 0.67 ± 0.06 Tyr (Y) 3.03 ± 0.02 3.50 ± 0.04 3.58 ± 0.08 2.13 ± 0.15 Val (V) 6.73 ± 0.03 6.72 ± 0.06 4.01 ± 0.06 5.41 ± 0.44 Sources :
DisProt est une base de données dédiée aux protéines intrinsèquement non structurées. |
| 7. Liens Internet et références bibliographiques |
| "Introduction à la structure des protéines" (1996) Branden & Tooze - Ed. DeBoeck Université | |
|
Cours 1 : "Secondary structure and backbone conformation" (Cours "SwissProt") Cours 2 : "Super-secondary structure" (Cours "SwissProt") Cours 3 : "Tertiary Protein Structure and folds" (Cours "SwissProt") |
|
|
"Secondary structure of Proteins" Base de données PROWL : propriétés physico - chimiques des acides aminés, peptides, protéines. |
|
|
Les méthodes de prédiction de structures secondaires Chou & Fasman (1974) "Conformational parameters for amino acids in helical, beta-sheet, and random coil regions calculated from proteins" Biochemistry 13, 211 - 222 Chou & Fasman (1978) "Prediction of the secondary structure of proteins from their amino acid sequence" Adv. Enzymol. Relat. Areas Mol. Biol. 47, 45 - 147 Garnier et al. (1996) "GOR secondary structure prediction method version IV" Methods in Enzymology R.F. Doolittle Ed., vol 266, 540 - 553 Chen et al. (2006) "Improved Chou-Fasman method for protein secondary structure prediction" BMC Bioinformatics 7, S14 Zheng C & Lukasz K (2008) "Prediction of beta-turns at over 80% accuracy based on an ensemble of predicted secondary structures and multiple alignments" BMC Bioinformatics 9, 430 |
|
|
Dodd & Egan (1990) "Improved detection of helix-turn-helix DNA-binding motifs in protein sequences" Nucleic Acids Res 18, 5019 - 5026 Burkhard et al. (2001) "Coiled coils: a highly versatile protein folding motif" Trends Cell Biol. 11, 82-88 Pellegrini-Calace & Thornton (2005) "Detecting DNA-binding helix - turn - helix structural motifs using sequence and structure information" Nucleic Acids Res 33, 2129 - 2140 Vacic et al. (2007) "Composition Profiler: A tool for discovery and visualization of amino acid composition differences" BMC Bioinformatics 8, 211 |
|
![]()