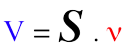| Métabolomique - Modèles de reconstruction métabolique à l'échelle d'un génome |
| Tweet |
|
|
1. Le métabolome et la métabolomique
2. Modèles de reconstruction du métabolisme à l'échelle d'un génome
3. Données et outils pour l'élaboration de modèles de reconstruction métabolique 4. La méthode de l'analyse de l'équilibre des flux pour l'obtention de modèles de reconstruction métabolique
|
5. Autres méthodes pour le développement de modèles de reconstruction métabolique 6. Démarche globale de la reconstruction métabolique à l'échelle d'un génome
7. Illustration : étude du contrôle du métabolisme glucidique lié au diabète de type 2 8. La base de données de modèles : "Biomodels" - EBI 9. Liens Internet et références bibliographiques |
|
1. Le métabolome et la métabolomique a. Introduction et définitions La métabolomique est définie actuellement comme l'analyse (identification, quantification, classification, découverte, ...) des petites molécules (masse molaire < 1000 - 1500 Da) constitutives d'une cellule, d'un tissu ou d'un fluide biologique. Ces molécules sont appelées métabolites et résultent de l'ensemble des réactions du métabolisme d'un organisme. Remarque : le métabolisme primaire et le métabolisme secondaire mettent en jeu beaucoup d'autres types de macromolécules. En particulier les enzymes, de même que des lipides et des oses dont les structures sont bien plus complexes et dont les masses molaires sont bien plus élevées.
Les métabolites sont très nombreux :
L'ensemble des métabolites constitue le métabolome :
Remarques :
|
Les investissements importants des gouvernements et des entreprises privées augmentent la croissance du marché de la métabolomique à l'échelle mondiale (intensifiée par le développement des techniques informatiques d'analyse des données de métabolomique). Le NIH ("National Institutes of Health") a annoncé un investissement de 14,3 millions de dollars en 2012 (et probablement 51,4 millions de dollars sur 5 ans) pour le développement de la métabolomique. Selon certains analystes, la demande mondiale en métabolomique :
Les grandes entreprises mondiales actuelle sont que Thermo Fisher Scientific Inc., Bruker Corporation, Agilent Technologies, Bio-rad Laboratories et Danaher Corporation. Elles collaborent activement avec les Universités et les laboratoires de recherche. Actuellement, les principaux champs d'investissements sont les biomarqueurs et la découverte de médicaments, la toxicologie, la nutrigénomique, la médecine personnalisée. |
| b. Incidences du métabolome sur les macromolécules biologiques et les processus cellulaires
La fonction de très nombreuses protéines est contrôlée par des métabolites. Par exemple :
Bien évidemment, l'activité des enzymes est contrôlée par les métabolites qui leur servent de substrat(s), de cofacteur(s) ou de coenzyme(s), d'inhibiteurs(s) ou d'effecteur(s) allostérique(s). Les métabolites influencent également le métabolisme des acides nucléiques :
Exemples de cofacteurs et coenzymes : coenzyme A, acide lipoïque, acide tétrahydrofolique, nicotinamide adénine dinucléotide (NAD+), flavine adénine dinucléotide, flavine mononucléotide, thiamine pyrophosphate, S-adénosylmethionine (SAM), S-adénosylcystéine, ... L'ATP illustre tout particulièrement le lien entre métabolites et processus cellulaires :
Enfin les métabolites ont une très forte incidence sur les propriétés physico-chimiques du milieu cellulaire, en priorité le pH (protons relargués par les divers métabolites acides et les réactions d'oxydo-réduction) que la cellule doit contrôler en permanence. Ainsi, la métabolomique permet d'analyser les rôles des métabolites dans la régulation de phénotypes. Par exemple, le NAD+ (coenzyme des oxido-réductases) est également un co-substrat, notamment des sirtuines (désacétylases dépendantes du NAD+), enzymes régulatrices des fonctions métaboliques et de la longévité. |
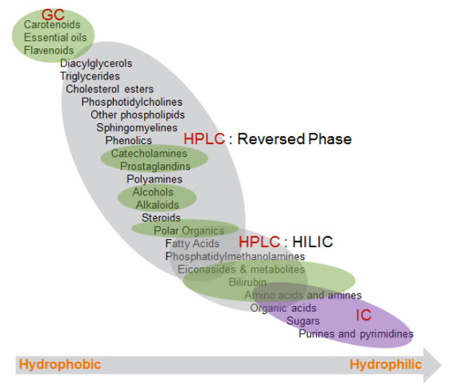
c. Techniques pour l'étude du métabolome Techniques physiques Les techniques actuelles ne permettent d'analyser que des métabolites de masse molaire limitée (quelques kDa). Les mieux adaptées à l'analyse du métabolome sont :
Source : Thermo Fisher Scientific Inc.
Métabolomique ciblée vs. non ciblée
Etude du métabolome avec des isotopes stables La variation du taux d'un métabolite donné résulte de la vitesse des enzymes qui le synthétisent et de celles qui l'utilisent comme substrat : cependant, les analyses métabolomiques ne fournissent pas d'informations sur le activités enzymatiques d'une voie métabolique. L'un des moyens de déterminer le rôle et le taux des métabolites dans les voies métaboliques consiste à suivre leur devenir avec des isotopes stables traceurs tels que le deutérium (2H), le carbone (13C) ou l'azote (15N). Analyse bioinformatique des données de métabolomique Outre la puissance accrue des calculateurs et la quantité de mémoire quasi illimitée des serveurs, l'identifications des métabolites est facilitée par des bases de données de métabolites telles que :
Des ressources sont mises à la disposition des laboratoires non équipés ou ne disposant pas des compétences requises en métabolomique. Par exemple :
|
2. Modèles de reconstruction du métabolisme à l'échelle d'un génome De plus en plus de génomes sont séquencés ou en cours de séquençage. Voir les bases de données suivantes :
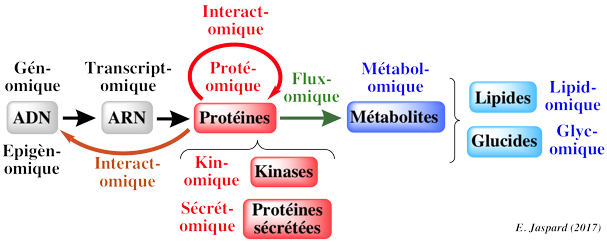
Les modèles du métabolisme à l'échelle d'un génome établissent le lien entre les informations issues du séquençage de ce génome (et les informations issues des analyses subséquentes de ce génome), les données biochimiques et métaboliques et les phénotypes liés à ce métabolisme. Cette démarche est récente car elle est la conséquence logique de tous les domaines antérieurs en "omique".
Quelques terminologies anglo-saxonnes :
|
b. Accroissement du nombre de modèles Le premier modèle de reconstruction du métabolisme à l'échelle d'un génome a été publié en 1999 pour Haemophilus influenzae (Edwards & Palsson, 1999). Pour l'être humain :
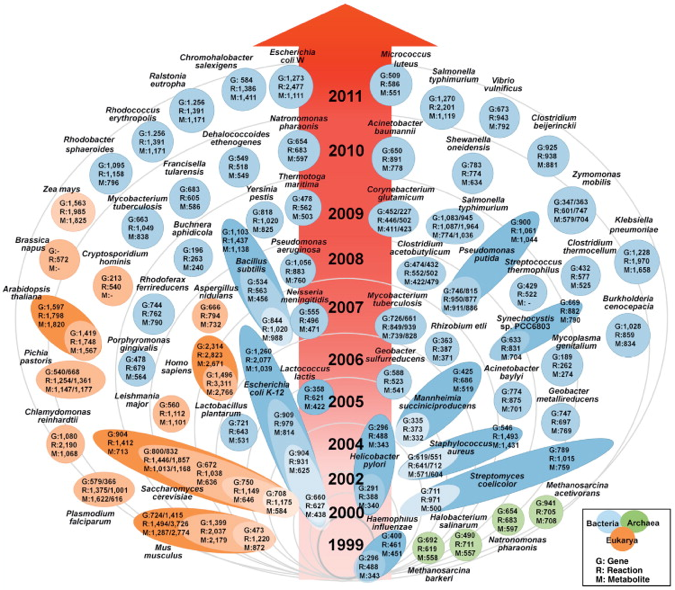
La figure ci-dessous montre le nombre croissant de modèles de reconstruction métabolique à l'échelle d'un génome.
Source : Kim et al. (2012) - orange : eukaryotes / bleu : prokaryotes / vert : archaea Le nombre de modèles croit exponentiellement : 30% des modèles ont été construits depuis 2010. Par exemple : la base de données dédiée au métabolome de E. coli (ECMDB) contient 3755 métabolites associées à 1402 enzymes, 387 transporteurs et 1542 voies métaboliques. Un nombre croissant de modèles d'Eucaryotes (beaucoup plus complexes) sont construits. Parmi eux, on note 3 plantes :
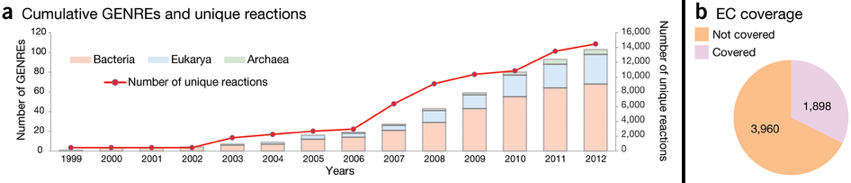
La figure ci-dessous montre le nombre cumulé de modèles du métabolisme à l'échelle d'un génome ("GEnome-scale Network REconstruction" - GENRE). La figure b indique le nombre d'enzymes annotées selon leur numéro EC ("Enzyme commission") intégrées dans les modèles.
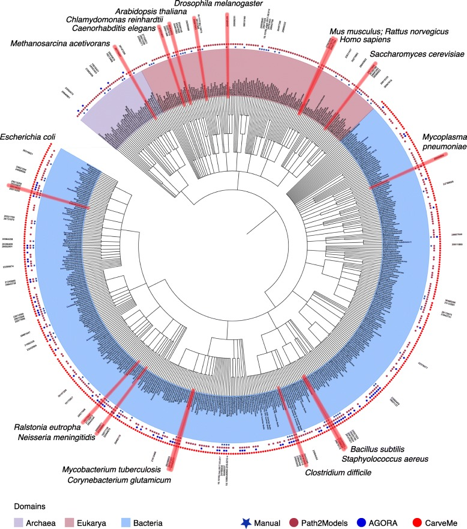
Source : Monk et al. (2014) Figure ci-dessous : arbre phylogénique des modèles métaboliques à l'échelle d'un génome de 6239 organismes (5897 bactéries en bleu clair, 127 archées en violet clair et 215 eucaryotes en rose - février 2019) :
Source : Gu et al. (2019) Voir : "Whole genome metabolism models available" - BioModels Database - EBI |
c. Les difficultés pour modéliser le métabolisme (liste non-exhaustive)
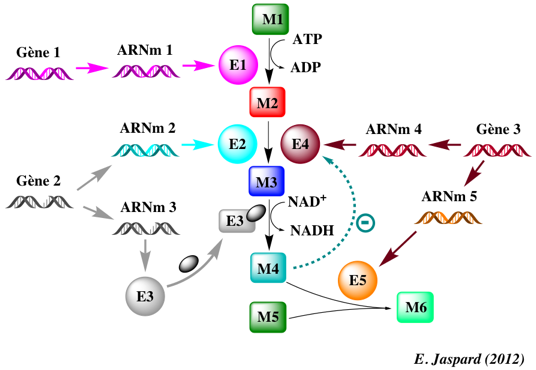
Figure ci-dessous : exemple (très simple en regard de la réalité biologique) qui illustre la complexité des phénomènes cellulaires
Cas des réactions et des enzymes manquantes (connues et/ou non connues)
k1 A -----> B k2 A -----> B + C
Gevorgyan et al. (2008) "Detection of stoichiometric inconsistencies in biomolecular models" Bioinformatics 24, 2245 - 2251 |
3. Données et outils pour l'élaboration de modèles de reconstruction métabolique La reconstruction du métabolisme à l'échelle d'un génome intègre l'ensemble des données issues (liste non exhaustive) :
Les modèles de reconstruction du métabolisme à l'échelle d'un génome nécessitent donc de fouiller ("data mining") une quantité colossale de données et, bien souvent, de les nettoyer (les "curer" - curators). Ce travail méticuleux, extrêmement gourmand en temps est laborieux et systématique. Les nouvelles données, déduites de l'analyse de l'ensemble de ces informations de base, constituent donc "sur-information" :
Enfin et non des moindres :
Voir Durot et al. (2009) |
|
Il faut d'abord décrire l'ensemble des réactions : identifier les métabolites impliqués en déterminant leur stoechiomètrie et identifier les enzymes qui catalysent ces réactions. Les techniques de mesures quantitatives des métabolites les plus utilisées sont :
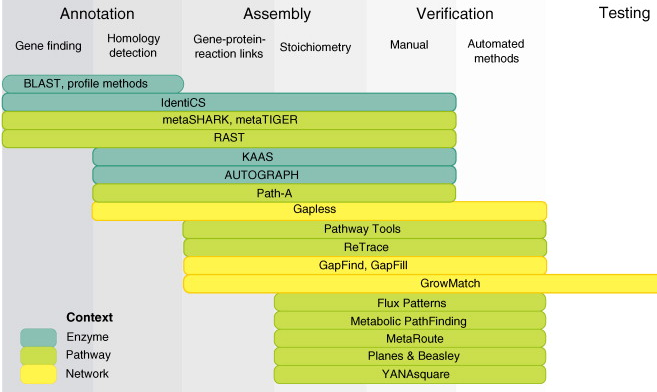
Les annotations fonctionnelles disséminées dans les bases de données doivent donc être compulsées, triées et les plus complètes possibles afin d'être traduites en un ensemble de réactions détaillées. La classification EC ("Enzyme Commission") de "Enzyme" fournit un système non ambigu d'association entre enzymes et réactions catalysées. Figure ci-dessous, exemples d'outils logiciels pour la reconstruction métabolique à l'échelle d'un génome. Cet exemple n'est qu'indicatif car ces outils évoluent très rapidement : de nouveaux outils de plus en plus performants sont régulièrement développés, d'autres (en ligne) ne sont pas maintenus et disparaissent.
Source : Pitkänen et al. (2010) |
| Exemples d'algorithmes / programmes pour la reconstruction métabolique à l'échelle d'un génome | |
| RFBA : "Regulatory Flux Balance Analysis" | EBA : "Energy balance analysis with Flux Balance Analysis" |
| SR-FBA : "Steady-state Regulatory Flux Balance Analysis" | TMFA : "Thermodynamics-based Metabolic Flux Analysis" |
| GapFind/GapFill | GrowMatch : "An Automated Method for Reconciling In Silico/In Vivo Growth Predictions" |
| GeneForce : "An Automated Phenotype-Driven Approach (GeneForce) for Refining Metabolic and Regulatory Models" | SMILEY algorithm |
| TIGER : "Toolbox for integrating genome-scale metabolic models, expression data, and transcriptional regulatory networks" | RAVEN toolbox (suite logicielle pour Matlab) |
| Exemples de bases de données pour la reconstruction métabolique à l'échelle d'un génome | |
| GOLD ("Genomes OnLine Database") : base de données de tous les génomes séquencés ou en cours de séquençage | CMR ("Comprehensive Microbial Resource") : ensemble d'informations et de programme pour l'analyse des génomes de procaryotes |
| GeneCards : base de données de gènes de l'homme | YMDB : "The Yeast Metabolome Database" |
| Enzyme : base de données de nomenclature des enzymes avec les N° EC associés | Brenda : base de données d'enzymes |
| TCDB ("Transporter Classification Database") : protéines de transport membranaires | BioCyc (contient les sous-bases de données EcoCyc (Escherichia coli K-12), AraCyc (Arabidopsis thaliana) , ...) : voies métaboliques et programmes pour les analyser |
| Pubchem : base de données de métabolites | metaTIGER : base de données de voies métaboliques et d'informations phylogénomiques pour un trés grand nombre d'eucaryotes |
| IntAct : base de données d'interactions entre protéines | STRING : "Known and Predicted Protein-Protein Interactions" |
| The Model SEED : "A resource for the generation, optimization, curation, and analysis of genome-scale metabolic models" | MetaRoute : "web interface for interactive navigation through genome-scale networks and local network visualization" |
| ERGO : "Comparative analysis of genomes and generation of sophisticated metabolic and cellular reconstructions" | KEGG ("Kyoto Encyclopedia of Genes and Genomes") : base de données pour l'étude des systèmes biologiques |
| Gene Ontology, DAVID , ... : annotation | KAAS (KEGG Automatic Annotation Server) : annotation |
| Les grandes bases de données généralistes : NCBI, EBI, Uniprot, PFAM, PDB, ... | |
Des langages informatiques sont également développés pour générer des fichiers de format standardidés aussi bien pour leur emploi par différentes programmes que pour comparer différents modèles de rcnstruction. Le langage le plus communément employé est SBML ("Systems Biology Markup Language"). |
4. La méthode de l'analyse de l'équilibre des flux pour l'obtention de modèles de reconstruction métabolique L'analyse de l'équilibre des flux ne nécessite pas de connaitre la concentration des métabolites ni les vitesses des réactions enzymatiques du système métabolique étudié. La question s'énonce de la manière suivante : compte-tenu des nutriments disponibles, quel ensemble de flux métaboliques maximise la croissance de l'organisme tout en maintenant la concentration intracellulaire des métabolites ?
|
b. Formalisme des vitesses des réactions du métabolisme Le formalisme des vitesses des réactions enzymatiques peut-être une expression complexe de [la concentration des métabolites x constantes de vitesse des réactions]. De plus ces vitesses sont en permanence modulées par les processus de régulation :
Décrire par le détail les équations qui traduiraient ces deux parties (cinétique enzymatique et processus de régulation) est une gageure tant sur le plan mathématique qu'en ce qui concerne le nombre de données requises. Et ce d'autant que l'on a aucune certitude de disposer de manière exhaustive de ces données. Pour s'en convaincre, il suffit de regarder ce que l'on connait du métabolisme de base. On mesure la complexité inouie des modèles qu'il faudrait développer. En pratique, à ce jour, on restreint les modèles sur le plan cinétique des réseaux métaboliques d'une cellule à des modèles plus modestes qui incluent des miliers de réactions (d'enzymes) et de métabolites, associés à autant (voire davantage) de gènes. |
|
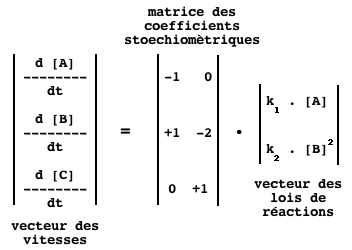
Pour l'ensemble du système, on a la relation :
Qui se traduit, dans le cas du système des réactions R1 et R2, par le formalisme :
|
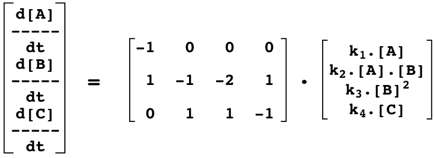
|
Qui se traduit par le formalisme :
|
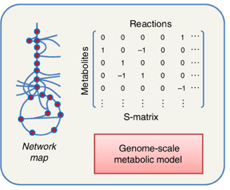
e. La matrice de stoechiométrie S ("S-matrix" ou "Stoichiometric matrix") Elle permet d'établir une relation linéaire, donc mathématiquement simple, entre :
Par convention :
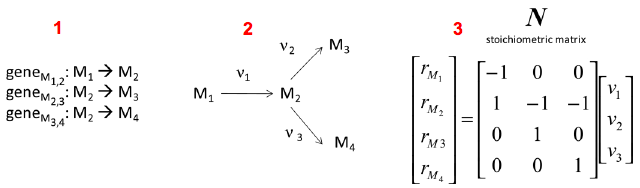
Source : Durot et al. (2009) Exemple simple de la construction d'une matrice de stoechiométrie N : on établit une liste de gènes qui codent les enzymes qui catalysent les réactions ν1, ν2 et ν3 impliquant les métabolites M1, M2, M3 et M4 (partie i figure ci-dessous).
Source : Weckwerth (2011)
|
Remarque Modéliser l'ensemble des évènements intra- et extra-cellulaires, surtout chez les Eucaryotes, est donc d'une extrême compléxité. Les modèles actuels ne tiennent pas (encore) compte de tous les mécanismes qui régulent le métabolisme et, à une plus grande échelle, l'ensemble des processus cellulaires. Quelques exemples traduisent la complexité des paramètres dont il faut tenir compte pour essayer de "simuler" une cellule in silico :
A cela s'ajoute les limites (toujours repoussées) qu'imposent la puissance de stockage et de calcul des ordinateurs. |
|
La notion d'état stationnaire est capitale. Cette approximation est justifiée en regard d'échelles de temps trés différentes :
Dès lors :
En conséquence, afin de simplifier les modèles :
Ainsi, à l'état stationnaire : α. Pour chaque métabolite d'un tel système "équilibré", on peut écrire une expression mathématique simple de la contrainte d'équilibre des masses qui met en jeu la vitesse des réactions : ∑ Si . νi = 0
Exemple :
β. La vitesse de croissance de la biomasse de la cellule (νbiomasse) doit être maximisée : z = cT.ν
Remarques :
|
|
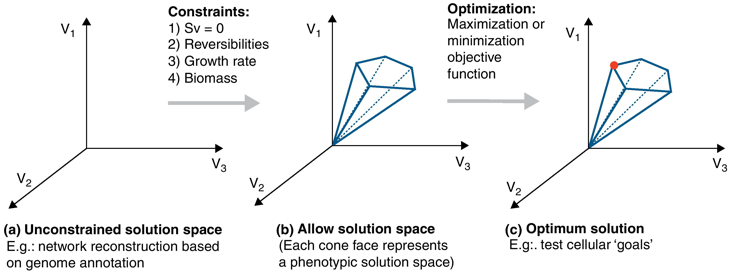
Figure a : en absence de contrainte, les valeurs des flux peuvent se trouver en tous points d'un espace de solutions.
Source : de Oliveira Dal'Molin & Nielsen (2013) Figure b : l'introduction de diverses contraintes restreint l'espace total des solutions à un sous-espace de solutions permises ou possibles (cône de la figure b).
En raison de ces contraintes, le flux global dans le réseau peut avoir toute valeur au sein de ce cône de solutions. Les valeurs en dehors de cet espace ne respectent pas la conservation de la matière et sont par conséquent éliminées. Figure c : en optimisant une fonction objectif, l'analyse de l'équilibre des flux permet de calculer une distribution de flux optimale unique qui se trouve aux bords de l'espace des solutions admissibles (surface de Pareto). |
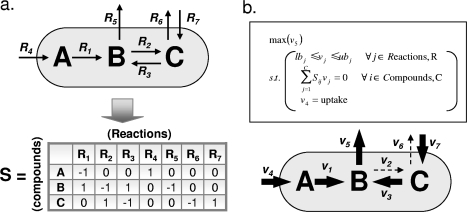
h. Exemples de fonctions objectif Les équations stoechiométriques contiennent davantage de flux inconnus que les équations d'équilibre des masses :
On émet donc des hypothèses quant au métabolisme : l'objectif le plus évident du métabolisme est une croissance maximale de l'organisme. Exemples de fonctions objectif implémentées dans des modèles de reconstruction sous contraintes (méthode "Flux Balance analysis" - FBA) :
Autres exemples de fonctions objectif :
Voir une liste de méthodes dite COBRA ("COnstraint-Based Reconstruction and Analysis"). |
i. La croissance et la biomasse Contraintes liées à la croissance de l'organisme Les organismes ont besoin de nutriments dont l'absorption (et l'excrétion) dépend de leur disponibilité, de leur concentration et de leur mode d'entrée dans l'organisme (métabolites à diffusion simple absorbés rapidement ou qui nécessitent un système de transport facilité ou actif). Si l'on détermine expérimentalement le taux d'absorption (et d'excrétion) d'un nutriment, cette donnée peut être inclue comme contrainte du flux limite du modèle métabolique : les nutriments absents ou non absorbés par l'organisme ne sont pas pris en compte dans son métabolisme (flux nul) et les taux d'absorption des nutriments présents sont correctement inclus dans la simulation. La biomasse Lors de la modélisation de la croissance d'un organisme, la fonction objectif est généralement définie comme la biomasse :
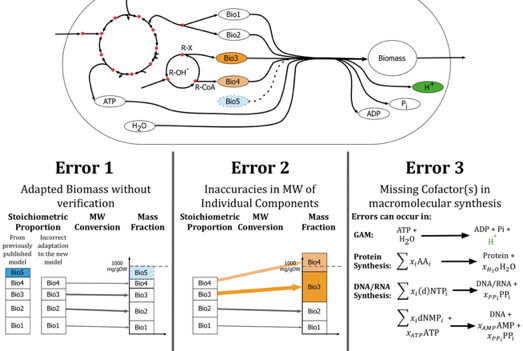
Il existe plusieurs sources d'erreurs dans l'analyse des réactions qui créent la biomasse :
Source : Chan et al. (2017)
|
|
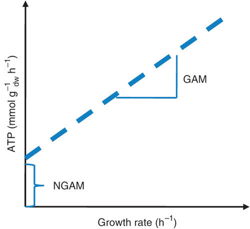
Les organismes effectuent des réactions cataboliques pour générer l'énergie nécessaire à leur croissance et à l'entretien des cellules. Cette énergie est traduite par 2 paramètres :
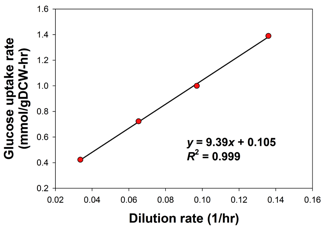
La GAM et la NGAM peuvent être déterminées directement avec le graphique des données de croissance obtenues à partir des expériences avec un chemostat.
Source : Thiele & Palsson (2010) Exemple : maximisation du renouvellement en ATP sous une contrainte d'absorption du glucose de 1 mmol.h-1.g-1 de matière sèche (figure ci-dessous).
Source : Chung et al. (2010)
Voir le package Python BOFdat qui propose des fonctions pour générer des coefficients stoechiométriques de fonction objectif de biomasse pour 5 types de macromolécules : ADN, ARN, protéines, lipides et métabolites (BOFdat à GitHub). |
| Approche | Régulation de la transcription non prise en compte | |
| Pas de coût d'enzyme | Avec coût d'enzyme | |
| Statique | Méthode FBA (1992) : optimiser une fonction objectif. Maintenir le système interne en état d'équilibre et optimiser un objectif donné par programmation linéaire. |
RBA ("Resource Balance Analysis" - 2001) : optimiser l'allocation des ressources pour maximiser le taux de croissance à l'état stationnaire. Modèles combinés : matrices ME (2012 - voir-ci-dessous) : optimiser un modèle métabolique et un modèle décrivant les processus cellulaires couplés. |
| Itérative | dFBA (SOA) ("dynamic FBA - Static Optimization Approach" - 1994) : optimiser plusieurs pas de temps de manière itérative. Diviser la période de temps étudiée en intervalles. Utiliser les conditions initiales ou les résultats du pas de temps précédent pour résoudre les équations dynamiques. Fixer les flux du modèle métabolique aux flux obtenus lors de la résolution des ODE. Optimiser le modèle au pas de temps suivant. |
Modèle dynamique : matrices ME (2019 - voir ci-dessus) : un modèle de l'organisme (noté "M-matrix") et un modèle de la stœchiométrie de synthèse des macromolécules (noté "E-matrix") sont combinés :
De plus, la biomasse est ajustée pour refléter les coûts de la synthèse des macromolécules et des acides aminés car les protéines peuvent s'accumuler dans ce modèle. |
| Dynamique | dFBA (DOA) ("dynamic FBA - Dynamic Optimization Approach" - 2002) : optimiser plusieurs pas de temps simultanément. Optimisation simultanée sur toute la période de temps étudiée. |
|
| Approche | Prise en compte de la régulation de la transcription | |
| Pas de coût d'enzyme | Avec coût d'enzyme | |
| Statique | SR-FBA ("Steady-state Regulatory FBA" - 2007) PROM ("Probabilistic Regulation Of Metabolism" - 2010) : utilisation de données de puces à ADN ("microarray") pour définir la limite supérieure des réaction à un certain pourcentage de la limite supérieure maximale. |
----- |
| Itérative | rFBA ("regulatory FBA" - 2001) : optimiser un réseau métabolique et de régulation. iFBA ("integrated FBA" - combinaison des méthodes dFBA-SOA et rFBA - 2008) : optimiser un réseau métabolique, un réseau régulation et un réseau cinétique intégrés. |
idFBA (2008) : optimiser un réseau [métabolique, de régulation et de signalisation] intégré. Méthode dFBA-SOA avec des données de phénotype supplémentaires. Après chaque pas de temps, les variables liées au phénotype sont mises à jour. Contraintes du pas de temps suivant avec ces variables mises à jour. |
| Dynamique | ----- | r-deFBA ("regulatory dynamic enzyme-cost FBA" - 2019) : optimiser l'utilisation des ressources cellulaires et la régulation de la transcription dans un cadre [discret-continu] hybride. Prédiction d'états régulateurs discrets corrélés à la dynamique continue des flux des réactions, des apports externes en substrats et enzymes et des protéines régulatrices nécessaires pour atteindre un objectif cellulaire tel que la maximisation de la biomasse sur un intervalle de temps. |
| Méthode | Description | |
| TMFA ("Thermodynamics-Based Metabolomic Flux Analysis") | Intégration des contraintes thermodynamiques dans le réseau métabolique | Modification de la réversibilité des réactions en fonction de leur variation d'énergie libre. |
| QQSPN ("Quasy-steady state Petri nets") | Optimiser un réseau [métabolique, de régulation et de signalisation] intégré à l'aide d'un réseaux de Petri | Calculer la contrainte et les nœuds objectifs pour chaque pas de temps. Mettre à jour le modèle métabolique en fonction de l'étape précédente. Optimiser le modèle métabolique. Mettre à jour les nœuds objectif du réseaux de Petri en fonction du nouvel objectif. |
Acronymes (modèles cinétiques et thermodynamiques) : CoCCoA : concentration change coupling analysis; GMA : generalized mass action; Lin-Log : linear in logarithms; Log-Lin : logarithmic-linear; MASS : mass action stoichiometric simulation; MCA : metabolic control analysis; MRL : modular rate law; ORACLE : optimization and risk analysis of complex living entities. |
||
Les langages pour coder les modèles Des normes d'encodage des modèles ont été créées pour échanger et reproduire ces modèles. La norme pour les modèles de biologie des systèmes est SBML (Hucka et al., 2003) :
Les éléments centraux de SBML sont utilisés pour écrire les modèles mathématiques des réseaux biologiques basés sur les réactions et permettent de coder des modèles informatiques basés sur les ODE. Il existe d'autres normes :
Outils de programmation : DFBAlab (code MATLAB pour dFBA), COBRA, DyMMM, ... |
6. Démarche globale de la reconstruction métabolique à l'échelle d'un génome C'est une démarche itérative : la comparaison avec la réalité biologique permet d'ajuster le modèle puis de confronter de nouveau celui-ci aux données réelles et ainsi de suite, afin d'améliorer ce modèle (voir la figure 1 de Balagurunathan et al., 2012).
Source : Lewis et al. (2012)
Source : Lewis et al. (2012) La reconstruction métabolique à l'échelle d'un génome s'appuyant massivement sur des méthodes bioinformatiques, elle est trés largement prédictive. Mais elle permet d'orienter de futures expériences dans des voies prometteuses et épargne un temps précieux de recherche au hasard. Bien évidemment, il existe une étape clé: la confrontation d'un modèle avec la réalité biologique. Cette étape est d'autant plus importante que les modèles peuvent être modifiés, améliorés comme tout système d'apprentissage. |
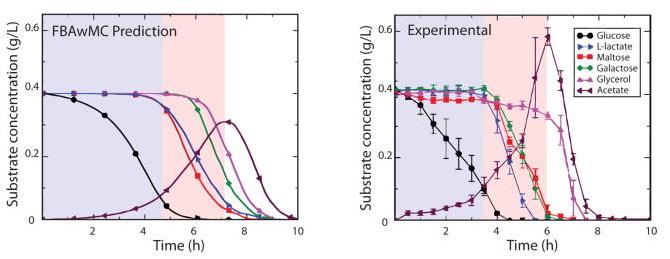
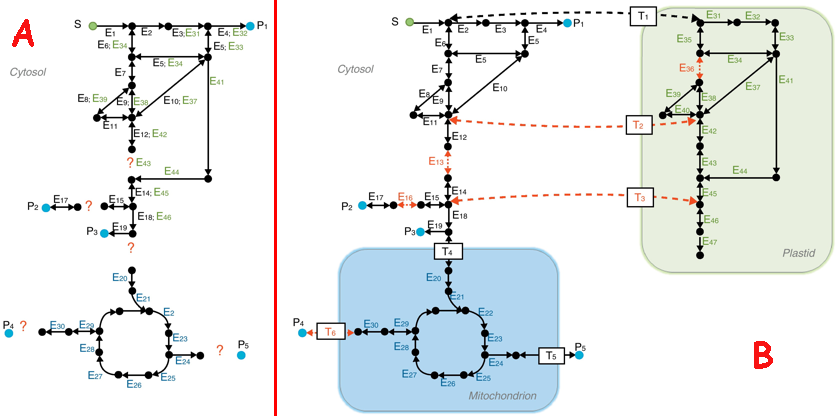
b. Amélioration d'un modèle de reconstruction métabolique Figure A (ci-dessous) : le 1er modèle dit "à plat"
Source : Gomes de Oliveira Dal'Molin & Nielsen (2013) Figure B : amélioration du 1er modèle
|
Exemple d'outil de contrôle de la qualité des modèles métaboliques La suite de tests nommée MEMOTE ("MEtabolic MOdel TEsts") est une méthode de contrôle de la qualité des modèles métaboliques. C'est un programme (en Python) :
|
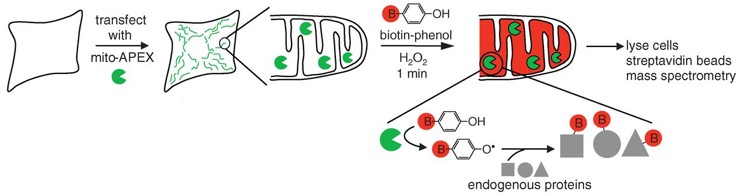
c. Illustration de l'amélioration d'un modèle de reconstruction via celle de l'annotation Les méthodes traditionnelles pour les études protéomiques d'organites se traduisent souvent par une spécificité limitée des protéines étudiées, la perte de matériel biologique et d'éventuelles contaminations du fait des étapes d'isolement des organites et de purification de leur contenu en protéines. Une méthode récente permet de minimiser ces inconvénients : elle utilise un marquage spécifique des protéines de la matrice de la mitochondrie par la biotine, tandis que la cellule est vivante, avec toutes ses membranes et ses complexes protéiques intacts et que les relations spatiales entre les protéines sont préservées. La méthode utilise une enzyme, l'ascorbate peroxidase (APEX), modifiée par ingénierie. L'APEX est spécifiquement adressée à la matrice mitochondriale par fusion à un peptide d'adressage de 24 acides aminés. Une fois dans la matrice mitochondriale, l'APEX marque par biotinylation (liaison covalente) les protéines voisines (mais pas les protéines lointaines) dans les cellules vivantes. L'APEX est active dans tous les compartiments cellulaires et elle oxyde de nombreux dérivés du phénol en radicaux phénoxyl. Ces radicaux :
Le marquage covalent est fait par l'addition de [biotine (B) - phénol] et de H2O2. Les cellules sont ensuite lysées et les protéines biotinylées sont récupérées avec des billes sur lesquelles est fixée la streptavidine. Les protéines sont ensuite éluées, séparées sur gel et identifiées par spectromètrie de masse.
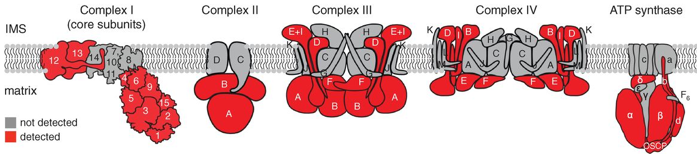
Source : Rhee et al. (2013) Cette étude a permis :
Source : Rhee et al. (2013) |
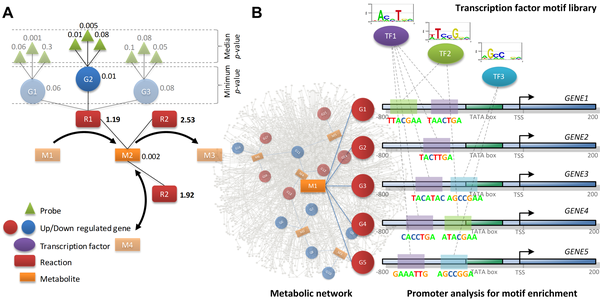
7. Illustration : étude du contrôle du métabolisme glucidique lié au diabète de type 2 Figure ci-dessous : schéma synoptique de la méthode d'identification de métabolites rapporteurs et des motifs de séquences régulatrices associées.
Source : Zelezniak et al. (2010) Figure A : Système de score pour l'identification de métabolites rapporteurs, basé sur les scores des réactions enzymatiques associées. Pour chaque enzyme, un score est aussi calculé sur la base de la "p-value" de la séquence nucléotidique du gène correspondant. Dans le cas de réactions catalysées par un complexe enzymatique ou un ensemble d'isoformes d'une même enzyme, la valeur minimale de la "p-value" de ces enzymes est retenue.
Figure B : Identification des motif de fixation des facteurs de transcription. Pour un métabolite rapporteur, un ensemble de gènes homologues codant des enzymes qui sont sur- ou sous-exprimés est sélectionné. Les régions promotrices et les motifs situés en amont des sites de démarrage de la transcription ("transcription start site - TSS") de chaque gène sélectionné sont annotés selon leur teneur en motifs de fixation des facteurs de transcription ("TF"). Signatures métabolique et régulatrice des diabètes de type 2
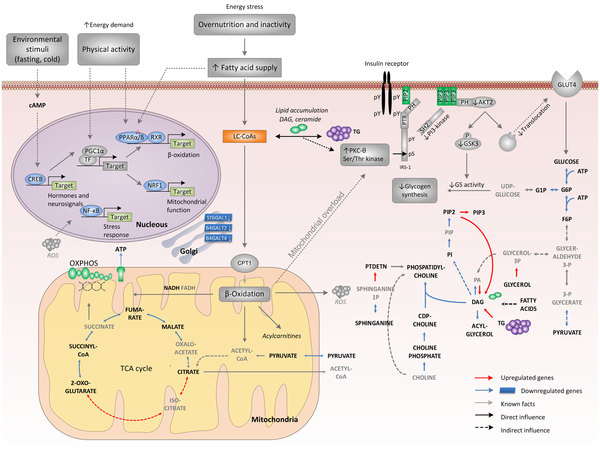
Source : Zelezniak et al. (2010) Légende : CDP-choline, cytidine diphosphate choline; G1P, glucose 1-phosphate; G6P, glucose 6-phosphate; GSK3, glycogen synthase kinase-3; IRE1, inositol requiring kinase-1; LC-CoAs, long-chain acyl CoAs; PA, phosphatidate; PH, pleckstrin homology domain;PI, phospatidylinositol; PIP, phospatidylinositol 4-phospate; PIP2, phosphatidylinositol 4,5-bisphospate, PIP3, phospatidylinositol 3,4,5-trisphospate; PTB, phosphotyrosine binding domain; RXR, retinoid X receptor; SH2, src homology domain; TF, transcription factor; CPT1, carnitine palmitoyltransferase-1; PTDETN, phosphatidylethanolamine. Les métabolites rapporteurs appartenant aux voies clés de la régulation des diabètes de type 2 sont écrits en gras.
Une sur-alimentation chronique et un manque d'activité physique augmentent l'entrée d'acides gras, ce qui induit la β-oxidation via l'activation des gènes médiée par le facteur activé de prolifération des peroxysomes (récepteur nucléaire - PPARα/δ - "peroxisome proliferator-activated receptor alpha and delta") sans qu'il y ait une augmentation coordonnée du flux du cycle des acides tricarboxyliques (TCA). Une conséquence possible est l'accumulation dans les mitochondries de dérivés de métabolites (exemples : acylcarnitines ou molécules réactives dérivées de l'oxygène - "reactive oxygen species - ROS") issus d'une β-oxidation incomplète. Ces stress pourrait induire une "surcharge mitochondriale" qui, de concert avec des molécules lipidiques de signalisation (exemple : le dyacylglycérol - DAG) enclencheraient une cascade impliquant des (Ser/Thr) protéines kinases, cascade qui serait initiée par une nouvelle protéine kinase nPKCs ("novel protein kinase Cs"). Il en résulte une phosphorylation des sites (Ser/Thr) du substrat 1 du récepteur de l'insuline (IRS-1) qui a pour conséquences :
Ces 2 évènements entravent la translocation du transporteur du glucose de type 4 (GLUT4) ce qui diminue le transport du glucose et donc la synthèse du glycogène. Une augmentation de l'activité physique ou le jeûne activent la protéine PGC1α (co-activateur 1α du récepteur nucléaire PPARγ) et la protéine CREB ("cAMP response element binding protein"), un activateur puissant de PGC1. Ces évènements atténuent le effets du stress lipidique en augmentant le flux du TCA et en couplant l'activité induite par la fixation du ligand sur PPARα/δ avec le remodelage médié par PGC1α des voies métaboliques situées en aval telles que la respiration et la β-oxidation. Voir des applications en biotechnologies industrielles et médicales. |
|
8. La base de données de modèles biologiques "Biomodels" (EBI) Voir un cours sur la régulation du métabolisme.
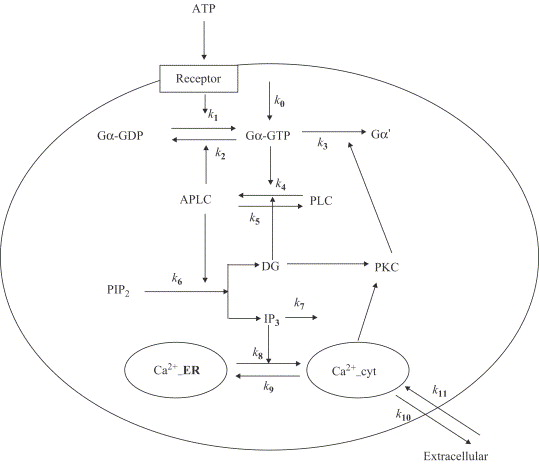
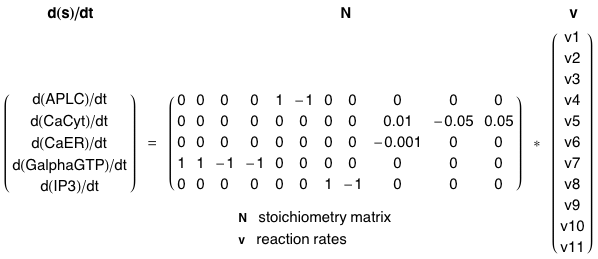
Figure ci-dessous : modèle proposé par Wang et al. (2007) - "A quantitative kinetic model for ATP-induced intracellular Ca2+ oscillations" J. Theor. Biol. 245, 510 - 519.
Variables du modèle :
|
a. Quel type de signal ce modèle essaye-t-il de modéliser ? b. Faire le lien entre les informations contenues dans l'onglet "Math" et le modèle décrit ci-dessus. c. Dans le menu déroulant "Actions" :
d. Quelle information obtient-on dans l'onglet «Physical entities» ? e. Aller à : "Models of the month".March 2010, model of the month - C. Hoyer : Borisov et al. (2009) "Systems-level interactions between insulin-EGF networks amplify mitogenic signaling" Mol Syst Biol. 5, 256 Quelques mots clés : "Epidermal growth factor / Insulin / EGF receptors (EGFR) / Insulin receptor / receptor tyrosine kinases" Cet article illustre le modèle : BIOMD0000000223 - Borisov 2009 EGF Insulin Crosstalk |
| 9. Liens Internet et références bibliographiques |
|
|
|
RECON 1, 2 et RECON 3D Rolfsson et al. (2011) "The human metabolic reconstruction Recon 1 directs hypotheses of novel human metabolic functions" BMC Systems Biology 5, 155 Thiele et al. (2013) "A community-driven global reconstruction of human metabolism" Nature Biotech. Swainston et al. (2016) "Recon 2.2: from reconstruction to model of human metabolism" Metabolomics 12, 109 Brunk et al. (2018) "Recon3D: A Resource Enabling A Three-Dimensional View of Gene Variation in Human Metabolism" Nat. Biotechnol. 36, 272 - 281 AGORA |
|
|
Edwards & Palsson (1999) "Systems properties of the Haemophilus influenzae Rd metabolic genotype" J. Biol. Chem. 274, 17410 - 17416 Hucka et al. (2003) "The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models" Bioinformatics 19, 524 - 531 Kell, DB (2004) "Metabolomics and systems biology: making sense of the soup" Curr. Opin. Microbiol. 7, 296 - 307 Alper et al. (2005) "Tuning genetic control through promoter engineering" Proc. Natl. Acad. Sci. USA 102, 12678 - 12683 |
|
|
Schuetz et al. (2007) "Systematic evaluation of objective functions for predicting intracellular fluxes in Escherichia coli" Mol. Syst. Biol. 3, 119 Henry et al. (2007) "Thermodynamics-Based Metabolic Flux Analysis" Biophys. J. 92, 1792 - 1805 Wang et al. (2007) "A quantitative kinetic model for ATP-induced intracellular Ca2+ oscillations" J. Theor. Biol. 245, 510 - 519 |
|
|
Oberhardt et al. (2008) Genome-Scale Metabolic Network Analysis of the Opportunistic Pathogen Pseudomonas aeruginosa PAO1" J Bacteriol. 190, 2790 - 2803 Lee et al. (2008) "Integrated dynamic Flux Balance Analysis (idFBA)" Gevorgyan et al. (2008) "Detection of stoichiometric inconsistencies in biomolecular models" Bioinformatics 24, 2245 - 2251 Lee JM et al. (2008) "Dynamic Analysis of Integrated Signaling, Metabolic, and Regulatory Networks" PLoS Comput Biol. 5 |
|
|
Oberhardt et al. (2009) "Applications of genome-scale metabolic reconstructions" Mol. Syst. Biol. 5, 320 Durot et al. (2009) "Genome-scale models of bacterial metabolism: reconstruction and applications" FEMS Microbiol Rev. 33, 164 - 190 Bennett et al. (2009) "Absolute Metabolite Concentrations and Implied Enzyme Active Site Occupancy in Escherichia coli" Nat. Chem. Biol. 8, 593 - 599 Borisov et al. (2009) "Systems-level interactions between insulin-EGF networks amplify mitogenic signaling" Mol Syst Biol. 5, 256 |
|
|
Zelezniak et al. (2010) "Metabolic Network Topology Reveals Transcriptional Regulatory Signatures of Type 2 Diabetes" PLOS Comput. Biol. 6, e1000729 Pitkänen et al. (2010) "Computational methods for metabolic reconstruction" Curr. Opin. Biotech. 21, 70 - 77 Thiele & Palsson (2010) "A protocol for generating a high-quality genome-scale metabolic reconstruction" Nat. Protoc. 5, 93 - 121 Yizhak et al. (2010) "Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model" Bioinformatics 26, i255 - i260 Chung et al. (2010) "Genome-scale metabolic reconstruction and in silico analysis of methylotrophic yeast Pichia pastoris for strain improvement" Microbial Cell Factories 9, 50 Barua et al. (2010) "An Automated Phenotype-Driven Approach (GeneForce) for Refining Metabolic and Regulatory Models" PLoS Comput Biol 6, e1000970 Jenkinson et al. (2010) "Thermodynamically consistent Bayesian analysis of closed biochemical reaction systems" BMC Bioinformatics 11, 547 Boghigian et al. (2010) "Utilizing elementary mode analysis, pathway thermodynamics, and a genetic algorithm for metabolic flux determination and optimal metabolic network design" BMC Syst Biol 4, 49 |
|
|
Weckwerth, W. (2011) "Unpredictability of metabolism - the key role of metabolomics science in combination with next-generation genome sequencing" Anal. Bioanal. Chem. 400, 1967 - 1978 Balagurunathan et al. (2012) "Reconstruction and analysis of a genome-scale metabolic model for Scheffersomyces stipitis" Microbial Cell Factories 11, 27 Kim et al. (2012) "Recent advances in reconstruction and applications of genome-scalemetabolic models" Curr. Opin. Biotechnol. 4, 617 - 623 |
|
Ebrahim et al. (2013) "COBRApy: COnstraints-Based Reconstruction and Analysis for Python" BMC Syst. Biol. 7, 74 Gomes de Oliveira Dal'Molin & Nielsen (2013) "Plant genome-scale metabolic reconstruction and modelling" Curr. Opin. Biotechnol. 24, 271 - 277 Lewis et al. (2013) "Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methods" Nat. Rev. Microbiol. 10, 291 - 305 |
|
|
Stanislav et al. (2014) "Progress toward single cell metabolomics" Curr. Opin. Biotechnol. 24, 95 - 104 Bordbar et al. (2014) "Constraint-based models predict metabolic and associated cellular functions" Nat. Rev. Genet. 15, 107-20 Monk et al. (2014) "Optimizing genome-scale network reconstructions" Nat. Biotechnol. 32, 447-452 |
|
|
Lin & Lin (2015) "Development of cell metabolite analysis on microfluidic platform" J. Pharm. Anal. 5, 337 - 347 Srinivasan et al. (2015) "Constructing kinetic models of metabolism at genome-scales: A review" Biotechnol. J. 10, 1345 - 1359 Johnson et al. (2016) "Metabolomics: beyond biomarkers and towards mechanisms" Nat. Rev. Mol. Cell Biol. 17, 451 - 459 do Rosario Martins Conde et al. (2016) "Constraint Based Modeling Going Multicellular" Front. Mol. Biosci. 3, 3 |
|
Magnusdottir et al. (2017) "Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota" Nat. Biotechnol. 35, 81 - 89 St. John et al. (2017) "Efficient estimation of the maximum metabolic productivity of batch systems" Biotechnol. Biofuels. 10, 28 Chan et al. (2017) "Standardizing biomass reactions and ensuring complete mass balance in genome-scale metabolic models" Bioinformatics doi: 10.1093/bioinformatics/btx453 |
|
Lachance et al. (2018) "BOFdat: generating biomass objective function stoichiometric coefficients from experimental data" doi: https://doi.org/10.1101/243881 Liu & Bockmayr (2019) "Regulatory dynamic enzyme-cost flux balance analysis: A unifying framework for constraint-based modeling" bioRxiv doi: 10.1101/802249v1 Gu et al. (2019) "Current status and applications of genome-scale metabolic models" Genome Biol. 20, 121 |
|
![]()