Les matrices de substitution pour la comparaison - alignement des séquences de nucléotides ou d'acides aminés |
| Tweet |
|
|
1. Les matrices pour les acides nucléiques 2. Calcul des valeurs élémentaires des matrices de substitution des acides aminés ("standard log-odds ratios matrices") 3. Les matrices protéiques
|
4. Liens Internet et références bibliographiques |
Voir un cours sur les algorithmes de comparaison et d'alignement de séquences. Plusieurs termes sont employés pour décrire une notion difficile : la « ressemblance » entre deux séquences biologiques :
Source : "Genet" R. Jalousot |
|
1. Les matrices pour les acides nucléiques Il existe peu de matrices pour les acides nucléiques car il n'y a que 4 lettres pour leur alphabet. La plus fréquemment utilisée est la matrice dite unitaire (ou matrice identité) où toutes les bases sont considérées comme équivalentes. |
| A | T | G | C | |
| A | 1 | 0 | 0 | 0 |
| T | 0 | 1 | 0 | 0 |
| G | 0 | 0 | 1 | 0 |
| C | 0 | 0 | 0 | 1 |
Matrice dite de transition - transversion |
| A | T | G | C | |
| A | 3 | 0 | 1 | 0 |
| T | 0 | 3 | 0 | 1 |
| G | 1 | 0 | 3 | 0 |
| C | 0 | 1 | 0 | 3 |
Matrice dite de BLAST |
| A | T | G | C | |
| A | 1 | - 3 | - 3 | - 3 |
| T | - 3 | 1 | - 3 | - 3 |
| G | - 3 | - 3 | 1 | - 3 |
| C | - 3 | - 3 | - 3 | - 3 |
2. Calcul des valeurs élémentaires des matrices de substitution des acides aminés ("standard log-odds ratios matrices") Ces matrices de substitution sont construites à partir de grands ensembles d'alignements de séquences de protéines, ces séquences ayant des fréquences d'acides aminés qu'on peu qualifier de "standard". Les alignements locaux de séquences (calculés de manière rigoureuse par l'algorithme de Smith-Waterman et de manière heuristique par BLASTP ou FASTA) nécessitent des matrices de scores qui génèrent en moyenne des valeurs négatives dans le cas de comparaison des séquences aléatoires. Si le score de matrice moyen ou attendu est positif, l'alignement s'étendra jusqu'aux extrémités des séquences et sera global plutôt que local. Les valeurs des matrices PAM (voir ci-dessous) ont été calculées comme le logarithme d'un rapport de probabilités (remarque : "log-odds" = "the logarithm of the odds" = le logarithme du rapport des probabilités) : fréquence d'alignement observée après une distance d'évolution donnée = homologues Les valeurs des matrices BLOSUM : même algèbre de rapport de probabilités mais les fréquences de transition ont été calculées en comptant le nombre de changements dans des blocs d'acides aminés dans des séquences de protéines apparentées (voir ci-dessous). |
Calcul numérique des valeurs de scores des matrices PAM et BLOSUM Dans le cas d'un modèle de protéine de séquence aléatoire (les acides aminés sont présents de manière indépendante les uns des autres, avec une fréquence individuelle f), toute matrice de scores (appropriée aux alignements locaux sans gap) avec au moins un score positif et un score attendu négatif (matrice "log-odds") peut être écrite sous la forme : Sij = (1/λ) . log (pi,j / fi . fj) Sij est le score élémentaire calculé pour l'alignement de 2 acides aminés i et j :
Avec les paramètres :
|
Exemple 1 :
Exemple 2 :
Le score de bit (S') est dérivé du score d'alignement brut (S) en tenant compte des propriétés statistiques du système de calcul des scores (en particulier la matrice de score et la taille de la base de données interrogée). Puisque les scores de bit sont normalisés par rapport au système de calcul des scores, ils sont utilisés pour comparer les scores de différents alignements. |
Entropie relative Toute matrice, quels que soient son type et son indice, est caractérisée par son contenu en information, appelée entropie relative ("relative entropy H : average mutual information per amino acid pair") qui est liée au nombre de résidus d'acides aminés qui doivent être alignés pour obtenir un score significatif statistiquement.
De manière schématique, on peut faire les approximations suivantes en considérant une utilisation équi-probable des 4 nucléotides (p = 0,25) et des 20 acides aminés (p = 0,05), ce qui ne reflète pas la réalité biologique. Nucléotides
Acides aminés
|
3. Les matrices protéiques |
| séquence 1 | Y | K | Y |
| séquence 1 | Y | K | C |
| Acides aminés i | Y | C | K |
| substitutions observées : ∑ jAij | 1 | 1 | 0 |
| fréquence d'apparition : fi | 3 | 1 | 2 |
| Mutabilité : mi | 0,33 | 1 | 0 |
Calcul des scores et normalisation
|
| Quelques matrices de scores de substitution des acides aminés | |
| Auteurs (année) | Matrice - principe de construction |
| Fitch & Margoliash (1967) | "minimum base change matrix for amino acid exchange converted to similarity measure" |
| Dayhoff et al. (1978) | matrices PAM (voir ci-dessous) |
| McLachlan (1971) | matrice dérivée de 16 familles de protéines |
| Grantham (1974) | matrice dérivée de trois propriétés physico-chimiques des acides aminés |
| Doolittle (1979) | "intuitive structural-genetic matrix" |
| Miyata et al. (1979) | matrice dérivée de la polarité et du volume moléculaire des acides aminés (Grantham, 1974) |
| Levin et al. (1986) | matrice empirique & structures secondaires |
| Rao (1987) | matrice dérivée des paramètres de Chou & Fasman (1974) |
| Risler et al. (1988) | matrice dérivée de la comparaison des structures 3D de 11 familles de protéines homologues |
| Gonnet et al. (1992) | matrices Gonnet (voir ci-dessous) |
| Henikoff & Henikoff (1992) | matrices BLOSUM (voir ci-dessous) |
| Jones et al. (1992) | matrices MS dérivées de 23.000 séquences de protéines |
| Johnson & Overington (1993) | matrices JOHM - substitutions dans des parties similaires des structures de protéines |
| Jones et al. (1994) | matrices JTT |
| Ng et al. (2000) | matrices PHDhtm |
| Kann et al. (2000) | matrice OPTIMA |
| Muller et al. (2002) | matrices VTML (voir ci-dessous) |
| Midic et al. (2009) | matrices MidicMat - régions désordonnées |
| Yamada & Tomii (2014) | matrices MIQS |
| Keul et al. (2017) | matrices PFASUM (voir ci-dessous) |
| Jia & Jernigan (2018) | matrices SeqStruct - corrélations [séquences / contacts au sein des structures de protéines] |
| Trivedi & Nagarajaram (2019) | matrices EDSSMat - acides aminés des régions désordonnées des protéines eucaryotes |
b. Les matrices PAM ("Point Accepted Mutation") Elles ont été créées par Margaret Dayhoff et ses collaborateurs, après l'alignement d'environ 1300 séquences très semblables (> 85% d'identité) appartenant à 71 familles de protéines.
Source : M. Dayhoff (1925-1983). Cette photo est la propriété de sa fille R. Dayhoff et mise à disposition par la National Library of Medicine. Les matrices PAM donnent la probabilité que, suite à une mutation par substitution au cours de l'évolution, n'importe quel acide aminé remplace n'importe quel autre acide aminé sans que la fonction de la protéine ne soit altérée, d'où la terminologie "mutation acceptée". Principe de construction des matrices PAM Elles sont construites avec des modèles de chaînes de Markov :
A chaque matrice XPAM correspond une matrice PAMX, appelée matrice de mutation de Dayhoff : ce sont les matrices PAMX qui sont utilisées par les algorithmes d'alignement. Voir le détail mathématique de la construction des matrices PAM. |
Exemple de la matrice PAM250 (ci-dessous) Cette matrice donne la probabilité que 250 mutations soit acceptées pour 100 acides aminés. Remarque : 0.99250 ≈ 0.08, ce qui signifie qu'après 250 multiplications avec un changement de 1%, environ 8% des acides aminés ne sont pas mutés. Du fait des mutations silencieuses et des réversions de mutations, cette matrice correspond à des séquences qui ont globalement 20% d'identité.
|
| A | R | N | D | C | Q | E | G | H | I | L | K | M | F | P | S | T | W | Y | V | |
| A | 2 | |||||||||||||||||||
| R | -2 | 6 | ||||||||||||||||||
| N | 0 | 0 | 2 | |||||||||||||||||
| D | 0 | -1 | 2 | 4 | ||||||||||||||||
| C | -2 | -4 | -4 | -5 | 4 | |||||||||||||||
| Q | 0 | 1 | 1 | 2 | -5 | 4 | ||||||||||||||
| E | 0 | -1 | 1 | 3 | -5 | 2 | 4 | |||||||||||||
| G | 1 | -3 | 0 | 1 | -3 | -1 | 0 | 5 | ||||||||||||
| H | -1 | 2 | 2 | 1 | -3 | 3 | 1 | -2 | 6 | |||||||||||
| I | -1 | -2 | -2 | -2 | -2 | -2 | -2 | -3 | -2 | 5 | ||||||||||
| L | -2 | -3 | -3 | -4 | -6 | -2 | -3 | -4 | -2 | 2 | 6 | |||||||||
| K | -1 | 3 | 1 | 0 | -5 | 1 | 0 | -2 | 0 | -2 | -3 | 5 | ||||||||
| M | -1 | 0 | -2 | -3 | -5 | -1 | -2 | -3 | -2 | 2 | 4 | 0 | 6 | |||||||
| F | -4 | -4 | -4 | -6 | -4 | -5 | -5 | -5 | -2 | 1 | 2 | -5 | 0 | 9 | ||||||
| P | 1 | 0 | -1 | -1 | -3 | 0 | -1 | -1 | 0 | -2 | -3 | -1 | -2 | -5 | 6 | |||||
| S | 1 | 0 | 1 | 0 | 0 | -1 | 0 | 1 | -1 | -1 | -3 | 0 | -2 | -3 | 1 | 3 | ||||
| T | 1 | -1 | 0 | 0 | -2 | -1 | 0 | 0 | -1 | 0 | -2 | 0 | -1 | -2 | 0 | 1 | 3 | |||
| W | -6 | 2 | -4 | -7 | -8 | -5 | -7 | -7 | -3 | -5 | -2 | -3 | -4 | 0 | -6 | -2 | -5 | 17 | ||
| Y | -3 | -4 | -2 | -4 | 0 | -4 | -4 | -5 | 0 | -1 | -1 | -4 | -2 | 7 | -5 | -3 | -3 | 0 | 10 | |
| V | 0 | -2 | -2 | -2 | -2 | -2 | -2 | -1 | -2 | 4 | 2 | -2 | 2 | -1 | -1 | -1 | 0 | -6 | 2 | 4 |
|
Les matrices PAM sont un peu moins utilisées maintenant au profit des matrices BLOSUM pour les raisons suivantes :
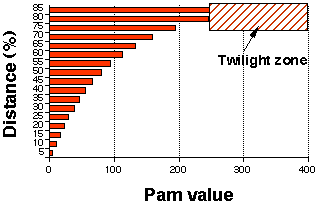
Correspondance entre la distance évolutive et la valeur des matrices PAM La zone d'ombre ("twilight zone") est la zone à partir de laquelle il devient difficile de dire si deux protéines sont homologues ou si elles se sont alignées par chance.
|
|
c. Les matrices BLOSUM ("BLOcks SUbstitution Matrix") Elles sont postèrieures aux matrices PAM et ont été développées par Henikoff & Henikoff en 1992.
LONM_YEAST|P36775 (632) GPPGVGKTSIGKSIARALNR 15 Calculs pour la construction des matrices BLOSUM
Exemple : la matrice BLOSUM62
Voir : "Interactive BLOSUM network visualization". Cet applet java permet de voir le lien entre la substitution d'un acide aminé par un autre et le % d'identité de la matrice et le score seuil de substitution. Matrices dérivées des matrices BLOSUM
|
d. Comparaison des matrices PAM et des matrices BLOSUM Les matrices PAM sont basées sur des modèles d'évolution explicites (c'est-à-dire que les substitutions sont comptabilisées à partir des valeurs des branches d'un arbre phylogénétique), tandis que les matrices BLOSUM sont basées sur des modèles d'évolution implicites. Les matrices PAM sont basées sur des mutations observées dans un alignement global, incluant aussi bien des régions hautement conservées que des régions hautement mutables. Les matrices BLOSUM sont basées uniquement sur des régions hautement conservées à partir d'alignements qui ne contiennent pas de brèches ("gaps"). La méthode pour comptabiliser les substitutions est différente. A l'inverse de la procédure suivie pour l'obtention des matrices PAM, celle des matrices BLOSUM utilise des groupes de séquences au sein desquels toutes les mutations n'ont pas le même poids, c'est-à-dire que les mutations ne sont pas toutes comptabilisées de manière identique.
Voir une comparaison des modes de calculs des valeurs des matrices PAM et BLOSUM. |
| Liens et différences entre PAM et BLOSUM | ||
| PAM | BLOSUM | |
| Pour comparer des séquences étroitement liées | des matrices avec des nombres plus faibles sont créées | des matrices avec des nombres plus élevés sont créées |
| Pour comparer des protéines distantes | des matrices avec des nombres élevés sont créées | des matrices avec des nombres faibles sont créées |
| Matrice | basée sur des alignements globaux de protéines étroitement apparentées | basée sur des alignements locaux |
| Un indice plus élevé dans la dénomination de la matrice reflète | une distance évolutive plus grande | une similarité de séquence plus élevée et donc une distance d'évolution plus petite |
| PAM1 est la matrice calculée à partir de comparaisons de séquences n'ayant pas plus de 15% de divergence mais correspondant à 99% d'identité de séquence. | BLOSUM 62 est la matrice calculée à partir de comparaisons de séquences avec une identité par paire non supérieure à 62%. | |
| d'autres matrices PAM sont calculées à partir de PAM1 | basé sur les alignements observés : ceux-ci ne sont pas extrapolés à partir de comparaisons de protéines étroitement apparentées | |
L'introduction de gaps dans les alignements réduit considérablement le contenu en information. L'effet est plus prononcé pour de faibles distances évolutives.
|
| Matrice | pénalité (ouverture / extension) de gap | Pourcentage de similarité | Information : bits/position | Nombre d'acides aminés pour un score statistiquement significatif (50 bits) |
| PAM70 | 10/1 | 33.9 | 0.58 | 86 |
| PAM30 | 9/1 | 45.9 | 0.90 | 56 |
| BLOSUM80 | 10/1 | 32.0 | 0.48 | 104 |
| BLOSUM62 | 11/1 | 28.9 | 0.40 | 125 |
| VTML140 | 10/1 | 28.4 | 0.44 | 114 |
| VTML120 | 11/1 | 32.1 | 0.54 | 93 |
| VTML80 | 10/1 | 40.5 | 0.74 | 68 |
| VTML40 | 13/1 | 64.7 | 1.92 | 26 |
| VTML20 | 15/2 | 86.1 | 3.30 | 15 |
| VTML10 | 16/2 | 90.9 | 3.87 | 13 |
| Source : How to select the right substitution matrix ? | ||||
|
Ce type de matrice a été construit en 1992 par Gonnet, Cohen et Benner. C'est une méthode itérative, sur la base de 16300 séquences de protéines correspondant à 2600 familles. Chaque séquence a été comparée à l'ensemble des séquences de la banque et les alignements ont été obtenus en utilisant une matrice initiale choisie arbitrairement. Une nouvelle matrice a été construite et les alignements ont été recalculés à partir de cette nouvelle matrice. Cette procédure a été répétée jusqu'à ce que la matrice reste inchangée. Différentes matrices Gonnet : Gonnet 40, Gonnet 120, ..., Gonnet 250, Gonnet 350. |
f. Exemples d'autres matrices et matrices spécialisées Matrices VTML (indices : 10, 20, 40, 80, 120, 140, 160, 200) Les matrices VTML ("Variable Time Maximum Likelihood ") ont été construites à partir d'un ensemble d'alignements de séquences 2 à 2 : les distances évolutives et les vitesses de substitution ont été estimées de manière itérative avec un estimateur de maximum de vraisemblance.
Les matrices qui ciblent les similarités faibles (exemples : BLOSUM45, PAM250 et VTML160) ont un contenu en information inférieur aux matrices qui ciblent les similarités élevées (exemples : BLOSUM90, PAM100 et VTML10). Illustration : un alignement a besoin de 50 bits (par exemple) pour être significatif du point de vue statistique.
Matrices PFASUM ("PFAm SUbstitution Matrix") C'est une série de matrices de substitution dérivée des alignements multiples des séquences "souches" de Pfam ("Pfam seed MSA" - version 29.0) qui couvrent la quasi totalité des séquences apparentées ou divergentes.
Voir un développement sur la base de données de familles de domaines protéiques Pfam. Autres matrices
Enfin, de plus en plus de matrices spécialisées sont développées. Elles s'appuient sur des jeux de données hautement spécifiques qui rassemblent des séquences (et d'autres informations comme des données de structure) de protéines ayant une relation structure - fonction particulière. Par exemple les protéines transmembranaires, en particulier les récepteurs couplés aux protéines G (ou RCPG). |
| Pourcentages des acides aminés dans différentes matrices de substitution et dans la base de données Swiss-Prot | |||||
| Acide aminé | GPCRtm |
JTTtm |
PHDhtm |
BLOSUM62 |
% issu des données |
| Ala (A) | 8.0 | 10.5 | 8.8 | 7.4 | 8.3 |
| Cys (C) | 3.6 | 2.2 | 2.6 | 2.5 | 1.4 |
| Asp (D) | 2.1 | 0.9 | 1.4 | 5.4 | 5.5 |
| Glu (E) | 1.9 | 1.0 | 1.0 | 5.4 | 6.7 |
| Phe (F) | 7.3 | 7.7 | 9.3 | 4.7 | 3.9 |
| Gly (G) | 4.6 | 7.6 | 5.7 | 7.4 | 7.0 |
| His (H) | 2.1 | 1.7 | 1.1 | 2.6 | 2.3 |
| Ile (I) | 8.1 | 11.9 | 11.0 | 6.8 | 5.9 |
| Lys (K) | 3.4 | 1.1 | 0.9 | 5.8 | 5.8 |
| Leu (L) | 14.1 | 16.3 | 16.0 | 9.9 | 9.7 |
| Met (M) | 3.1 | 3.3 | 4.1 | 2.8 | 2.4 |
| Asn (N) | 3.4 | 1.8 | 2.2 | 4.5 | 4.1 |
| Pro (P) | 3.8 | 2.6 | 3.2 | 3.9 | 4.7 |
| Gln (Q) | 2.2 | 1.4 | 1.2 | 3.4 | 3.9 |
| Arg (R) | 4.5 | 1.6 | 2.1 | 5.2 | 5.5 |
| Ser (S) | 6.8 | 5.7 | 6.5 | 5.7 | 6.6 |
| Thr (T) | 5.6 | 5.2 | 5.3 | 5.1 | 5.3 |
| Val (V) | 9.2 | 11.9 | 11.0 | 7.3 | 6.9 |
| Trp (W) | 1.9 | 2.2 | 1.9 | 1.3 | 1.1 |
| Tyr (Y) | 4.3 | 3.2 | 4.7 | 3.2 | 2.9 |
On remarque la proportion nettement plus importante d'acides aminés hydrophobes constitutifs des hélices transmembranaires. Ces chiffres évoluent (légèrement) au fur et à mesure que de nouvelles séquences sont ajoutées dans les différentes bases de données.
|
|||||
| 4. Liens Internet et références bibliographiques |
|
Astral Sequences & Subsets - SCOPe : jeux de données pour tester les performances des différents types de matrices BAliBASE : "A benchmark alignment database for the evaluation of multiple alignment programs" Théorie information - modélisation |
|
|
Shannon C.E. (1948) "A mathematical theory of communication" Bell Syst. Tech. J. 27, 379 - 423, 623 - 656 Dayhoff, Schwartz & Orcutt (1978) "A model of evolutionary change in proteins" dans "Atlas of protein sequence and structure", Dayhoff, M.O. (ed.), vol 5, 345 - 352 Johnson & Overington (1993) "A structural basis for sequence comparisons. An evaluation of scoring methodologies" J. Mol. Biol. 233, 716 - 738 Jones et al (1994) "A mutation data matrix for transmembrane proteins" FEBS Lett. 339, 269 - 275 |
|
|
Strait & Dewey (1996) "The Shannon information entropy of protein sequences" Biophys. J. 71, 148 - 155 Henikoff & Henikoff (1992) "Amino acid substitution matrices from protein blocks" Proc. Nat. Acad. Sci. USA 89, 10915 - 10919 Gonnet et al. (1992) "Exhaustive matching of the entire protein sequence database" Science 256, 1443-1444 Ng et al. (2000) "PHAT: a transmembrane-specific substitution matrix. Predicted hydrophobic and transmembrane" Bioinformatics 16, 760 - 766 Müller et al. (2002) "Estimating amino acid substitution models: a comparison of Dayhoff's estimator, the resolvent approach and a maximum likelihood method" Mol. Biol. Evol. 19, 8 - 13 |
|
|
Styczynski et al. (2008) "BLOSUM62 miscalculations improve search performance" Nat. Biotechnol. 26, 274 275 Song et al. (2015) "Parameterized BLOSUM matrices for protein alignment" IEEE/ACM Trans. Comput. Biol. Bioinform. 12, 686 - 694 Rios et al. (2015) "GPCRtm: An amino acid substitution matrix for the transmembrane region of class A G Protein-Coupled Receptors" BMC Bioinformatics 16, 206 |
|
|
Hess et al. (2016) "Addressing inaccuracies in BLOSUM computation improves homology search performance" BMC Bioinform. 17, 189 Keul et al. (2017) "PFASUM: a substitution matrix from Pfam structural alignments" BMC Bioinformatics 18, 293 Trivedi & Nagarajaram (2019) "Amino acid substitution scoring matrices specific to intrinsically disordered regions in proteins" Sci. Rep. 9, 16380 |
|
![]()