Algorithmes et programmes de comparaison de séquences Interprétation des résultats : E-value, P-value |
| Tweet |
|
|
1. Définitions 2. Algorithme de Needleman & Wunsch et algorithme de Smith & Waterman 3. Diversité des programmes - spécificité selon le type de données analysées 4. Programmes d'alignement local
|
5. Programme d'alignement multiple progressif : Clustal W 6. Modèle de Markov caché (Hidden Markov Model - HMM) 7. Interprétation des résultats : E-value, P-value 8. Liens Internet et références bibliographiques |
|
1. Définitions Il existe 3 grandes classes d'algorithmes de comparaison de séquences :
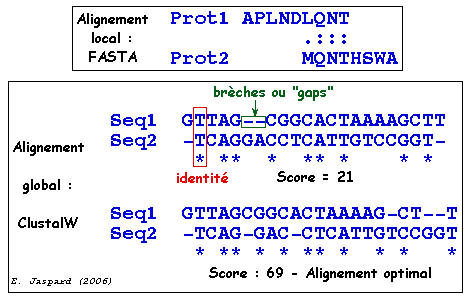
Alignement : processus par lequel deux (ou n) séquences sont comparées afin d'obtenir le plus de correspondances (identités ou substitutions conservatives) possibles entre les lettres qui les composent.
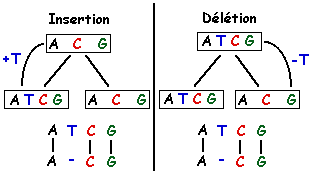
indel : in" = insertion et "del" = délétion
similarité : c'est le pourcentage d'identités et/ou de substitutions conservatives entre des séquences. Le degré de similarité est quantifié par un score. Le résultat de la recherche d'une similarité peut être utilisé pour inférer l'homologie de séquences.
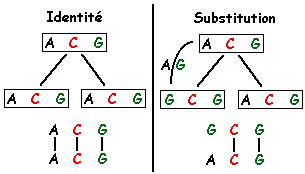
homologie : 2 séquences sont homologues si elles ont un ancêtre commun. L'homologie se mesure par la similarité : une similarité significative est signe d'homologie sauf si les séquences présentent une faible complexité. faible complexité ("low-complexity regions") : régions qui contiennent peu de caractères différents. Exemples : (a) FFFPPPPPVVV, 3 acides aminés différents seulement (région riche en proline) - queue poly-A des ARN. Ces régions posent des problémes dans l'analyse des séquences car elles génèrent un score biaisé. Exemple de programme qui analyse ce type de régions : "SEG". mésappariement : non correspondance entre deux lettres. Un mésappariement peut être :
score : un score global permet de quantifier l'homologie. Il résulte de la somme des scores élémentaires calculés sur chacune des positions en vis à vis des deux séquences dans leur appariement optimal. C'est le nombre total de "bons appariements" pénalisé par le nombre de mésappariements. score élémentaire :
|
|
Algorithme de Needleman & Wunsch |
Algorithme de Smith & Waterman |
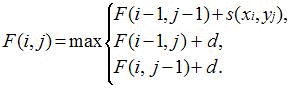 |
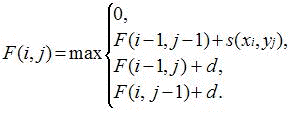
|
|
La ligne i = 0 et la colonne j = 0 sont initialisées aux valeurs de pénalité des gaps. La fonction de récurrence ne réinitialise pas la valeur à 0 si aucune valeur positive n'est présente. |
La ligne i = 0 et la colonne j = 0 sont initialisées à 0. N'importe quelle case de la matrice de comparaison peut être un point de départ pour le cacul des scores finaux. Si ce score devient inférieur à zéro, la fonction de récurrence réinitialise la valeur à 0 et la case peut être utilisée comme un nouveau point de départ. |
|
F(i,j) : valeur à la position (i,j) de la matrice. s(xi,yj) : valeur obtenue à partir de la matrice de substitution pour les nucléotides ou les acides aminés (xi,yj) correspondant à la position (i,j) de la matrice. C'est donc le score correspondant à l'alignement des lettres xi et yj. Ce score prend, par exemple, les valeurs suivantes :
Remarque : si on opte pour d'autres valeurs, on obtient d'autres alignements optimaux, d'où le choix crucial de la meilleure matrice de substitution lors des alignements. La fonction de pénalité d'un gap est définie par : f(n) = d + [e . (n-1)], où :
Exemple : un gap de longueur n = 3, avec une pénalité d'ouverture d = -10 et d'extension e = -2, aura un score de f(3) = -10 + (-2 x 2) = -14 |
Application : alignement de la séquence 1 = ACGCT avec la séquence 2 = ACT On remplit la 1ère ligne et la 1ère colonne de la matrice qui correspondent à un gap à la 1ère position :
F(1,1) aura pour valeur la valeur maximale de l'une des possibilités suivantes :
|
| j | 0 | 1 | 2 | 3 | |
| i | - (gap) | A | C | T | |
| 0 | - (gap) | 0 | -5 | -10 | -15 |
| 1 | A | -5 | 3 | -2 | -7 |
| 2 | C | -10 | -2 | 6 | 1 |
| 3 | G | -15 | -7 | 1 | 5 |
| 4 | C | -20 | -12 | -4 | 0 |
| 5 | T | -25 | -17 | -9 | -1 |
F(2,1) aura pour valeur la valeur maximale de l'une des possibilités suivantes :
Et ainsi de suite. Pour reconstituer l'alignement, on démarre de la dernière case (5,3) et on détermine la case à partir de laquelle cette case a été atteinte : a. la valeur -1 de la case (5,3) ne peut-être obtenue qu'en ajoutant +3 (soit une identité) à la valeur -4 [(case (4,2)]. Celà correspond à l'alignement du "T" de la séquence 1 avec le "T" de la séquence 2. b. la valeur -4 de la case (4,2) peut être obtenue de 2 manières :
c. Et ainsi de suite. Dés lors, on obtient 2 alignements optimaux qui ont le même score de +1. |
| séquence 1 | A | C | G | C | T |
| séquence 2 | A | - | - | C | T |
| séquence 1 | A | C | G | C | T |
| séquence 2 | A | C | - | - | T |
|
3. Diversité des programmes - spécificité selon le type de données annalysées Voir l'extrême diversité des programmes d'alignement et de comparaison de séquences. |
| Type de séquences | Protéines ou acides nucléiques (ADN et/ou ARN) ou les deux |
| Type d'alignement | Local ou global |
| Accessibilité | Serveur Web ou implémenté sur l'ordinateur (lignes de commandes) |
| Spécialisation de plus en plus prononcée du champs d'application des algorithmes / programmes |
|
| Les "benchmarks" sont de vastes ensembles de données (homogènes, curées, testées) qui permettent de comparer les performances d'algorithmes / programmes. | Exemples de "benchmarks":
|
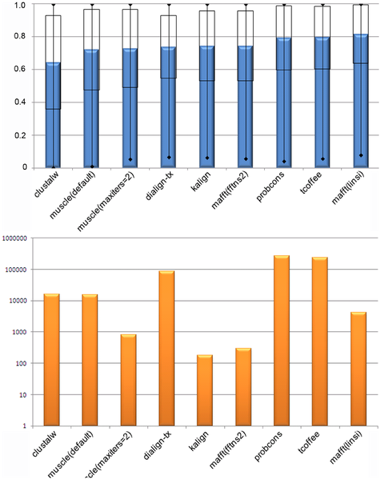
Figure ci-dessous : comparaison des performances de plusieurs programmes d'alignement de séquences
Source : Thompson et al. (2011)
Bleu : efficacité / Orange : rapidité (échelle log) |
| Programme | score d'efficacité | temps de calcul |
| Probcons | 79.4% | 2.7 jours |
| T-Coffee | 79.4% | 2.7 jours |
| Mafft (linsi) | 81.6% | 1.2 heures |
| Kalign | 74.3% | 3 minutes ! |
Les programmes sont de plus en plus spécifiques du type de données biologiques traitées ou du type d'analyse effectuée :
|
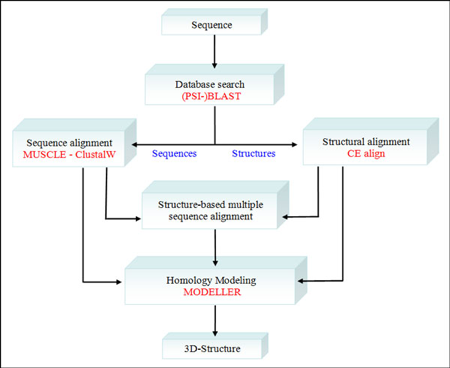
Illustration : la comparaison de structures et la modélisation par homologie On a de plus en plus d'informations qui tendent à démontrer que le nombre de repliements des protéines dans la nature est limité (quelques milliers). On peut donc regrouper les protéines selon le type de repliement qu'elles adoptent. Voir les bases de données CATH et SCOP, par exemple. Remarque : les protéines dites "intrinsèquement non structurées" sont à part. Le préalable de la modélisation par homologie ("homology modeling"- "protein threading") est de disposer d'au moins une protéine dont la structure 3D a été déterminée. Elle sert de "modèle" pour modéliser la structure 3D potentielle d'une protéine pour laquelle on ne dispose que de la séquence. Cette séquence doit bien sûr être proche (homologue) de celle de la protéine modèle. Il faut donc d'abord effectuer des alignements de séquences.
Figure ci-dessous : Procédure de "PyMod" qui intègre divers types de données et d'analyses :
Source : Bramucci et al. (2012)
Chaque "bloc de procédure" est indépendant des autres : on peut donc, par exemple, effectuer un alignement multiple de séquences sans recherche préalable dans une base de données. |
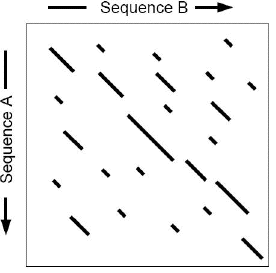
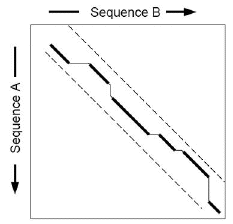
4. Programmes d'alignement local Les méthodes de programmation dynamique permettent de calculer, sous un système de scores donné, l'alignement optimal, global ou local, entre deux séquences en un temps proportionnel au produit des longueurs des deux séquences. Appliquées à une banque de séquences, le temps de calculs de ces méthodes augmente linéairement avec la taille de la banque. On définit 2 caractéristiques pour une méthode de comparaison de séquences :
Les programmes des familles Fasta et BLAST sont des heuristiques qui réduisent le facteur temps en "sacrifiant" un peu de sensibilité. L'un et l'autre simplifient le problème :
Ces étapes sélectives permettent :
Cette logique de recherche plus rapide dans son exécution, comporte donc le risque d'éliminer des séquences qui ont une similarité plus difficile à détecter ou d'aboutir à des alignements sub-optimaux. La sensibilité et la sélectivité se réfèrent à une notion de résultat significatif ou non. Les programmes mesurent une signification statistique des résultats par rapport à un modèle aléatoire : un résultat est considéré comme significatif si la probabilité de l'obtenir par hasard est trés faible. Les systèmes de score partent du postulat que les résultats les plus significatifs du point de vue statistique sont aussi les plus pertinents du point de vue biologique. Or ce n'est pas toujours le cas car des résultats biologiquement intéressants peuvent être non significatfs sur un plan statistique. En d'autres termes, la signification biologique d'une similarité entre des séquences n'est pas forcément estimable sur la seule valeur d'un score. |
|
b. Programme FASTA - Pearson & Lipman (1988) Le programme FASTA ne considère que les séquences présentant une région de forte similitude avec la séquence recherchée. Il applique ensuite localement à chacune de ces meilleures zones de ressemblance un algorithme d'alignement optimal. La codification numérique des séquences, c'est-à-dire la décomposition de la séquence en courts motifs (nommés uplets) transcodés en entiers, confère à l'algorithme l'essentiel de sa rapidité. Etape 1
Lorsqu'une séquence est comparée à une base de données, la première étape est effectuée pour chaque séquence présente dans cette base de données. Etape 2
Etape 3
Etape 4
Interprétation des résultats La sortie de FASTA se décompose en trois parties :
|
|
c. Le programme BLAST (et ses dérivés) - Altschul et al.(1990) Le programme BLAST (Basic Local Alignment Search Tool) s'appuie sur une méthode heuristique qui utilise la méthode de Smith & Waterman. C'est un programme qui effectue un alignement local entre deux séquences nucléiques ou protéiques. La rapidité de BLAST permet la recherche des similarités entre une séquence requête et toutes les séquences d'une base de données. Voir une description de l'algorithme de BLAST. Les différents programmes BLAST Acides nucléiques
|
|
Protéines
Altschul S. F. et al. (1997) "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs" Nucleic Acids Res. 25, 3389 - 3402 |
| Choix des différentes bases de données de séquences de protéines | |
| Bases de données | Description |
| nr | Non-redundant GenBank CDS translations + PDB + SwissProt + PIR + PRF, excluding those in env_nr. |
| refseq | Protein sequences from NCBI Reference Sequence project. |
| swissprot | Last major release of the SWISS-PROT protein sequence database (no incremental updates). |
| pat | Proteins from the Patent division of GenBank. |
| month | All new or revised GenBank CDS translations + PDB + SwissProt + PIR + PRF released in the last 30 days. |
| pdb | Sequences derived from the 3-dimensional structure records from the Protein Data Bank. |
| env_nr | Non-redundant CDS translations from env_nt entries. |
| Smart v4.0 | 663 PSSMs from Smart, no longer actively maintained. |
| Pfam v11.0 | 7255 PSSMs from Pfam, not the latest. |
| COG v1.00 | 4873 PSSMs from NCBI COG set. |
| KOG v1.00 | 4825 PSSMs from NCBI KOG set (eukaryotic COG equivalent). |
| CDD v2.05 | 11399 PSSMs from NCBI curated cd set. |
|
Les programmes FASTA et BLAST suivants sont équivalents :
|
|
PHI-Blast est un programme qui prend en entrée une séquence requête protéique et un motif défini par une expression régulière. PHI-Blast est adapté à la recherche de séquences protéiques qui contiennent un motif spécifié par l'utilisateur (fenêtre "PHI pattern" de la section "Algorithm") ET sont similaires à la séquence requête (fenêtre "Search") dans le voisinage proche du motif. La syntaxe du motif doit suivre la syntaxe de PROSITE.
Application :
|
|
Complément sur PSI-Blast PSI-Blast est adapté :
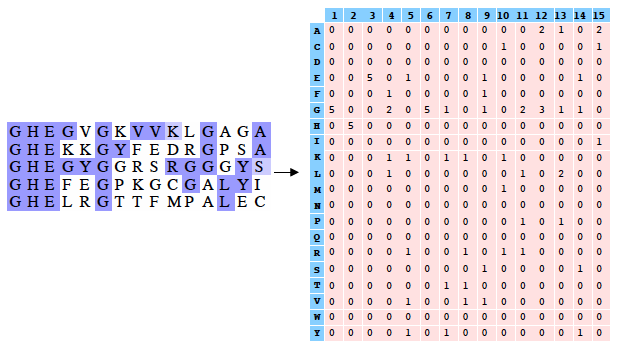
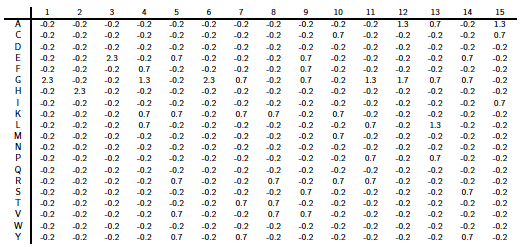
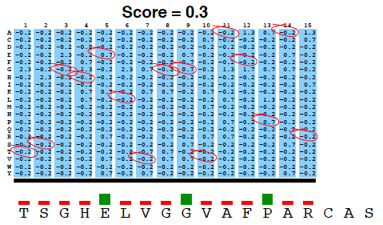
PSI-Blast construit un profil à partir de l'alignement multiple des séquences qui ont obtenu les meilleurs scores avec la séquence requête. Ce profil est comparé à la banque interrogée et est affiné au fur et à mesure des itérations. Ainsi, la sensibilité du programme est augmentée. Un profil est un tableau des fréquences observées des acides aminés (ou nucléotides) à chaque position dans un alignement multiple. Exemple (très simple) d'alignement multiple de 2 séquences de 4 acides aminés : DWKD Le profil de probabilités correspondant : 1 2 3 4 D 1.0 0.0 0.0 0.5 G 0.0 0.0 0.0 0.5 K 0.0 0.0 0.5 0.0 N 0.0 0.0 0.5 0.0 W 0.0 1.0 0.0 0.0 Ce qui ce signifie :
L'utilisation d'un profil permet une recherche beaucoup plus sensible de séquences homologues « éloignées » que l'utilisation d'une séquence seule car le profil contient de l'information sur la variabilité des différentes positions parmi les protéines connues. En contrepartie un profil est moins spécifique qu'une simple séquence seule. Si on utilise PSI-Blast sur un sous ensemble particulier de séquences, il est probable que l'on ne trouve pas tous les homologues, surtout si leur séquence est peu conservée par rapport à la séquence requête. Pour améliorer la sensibilité de la détection des homologues « éloignées », il est préférable d'effectuer un alignement avec PSI-Blast sur une banque de séquences plus grande. Mais la sensibilité est diminuée si la banque de données est trop grande puisque la fréquence d'observation d'un score particulier (la "E-value") augmente avec la taille de la banque de données. Or, pour un alignement de 2 séquences, plus le score est petit, plus la probabilité que ces 2 séquences soient homologues est grande. Il est donc préférable de chercher d'abord dans une banque "nettoyée" ("curated") comme la base de données non-redondante ("nr") où toutes les séquences identiques ont été éliminées sauf un exemplaire. Si plusieurs séquences sont dans cette banque, on peut calculer un profil et l'utiliser pour effectuer une nouvelle recherche dans ce sous ensemble. On augmente ainsi la sensibilité de la recherche d'homologues. Naumoff D.G. & Carreras M. (2009) "PSI Protein Classifier: a new program automating PSI-BLAST search results" Molecular Biology (Engl Transl) 43, 652 - 664 |
| Conclusion PSSM | |
|
Avantages
|
Inconvénients
|
Cette méthode est à utiliser pour modéliser de courtes régions avec une forte variabilité mais de longueurs constantes. Outils :
Bases de données : |
|
Application à PSI-Blast 1. Une recherche standard BLAST est effectuée contre une base de données en utilisant une matrice de substitution. 2. Une matrice PSSM est construite automatiquement à partir d'un alignement multiple des séquences ayant le plus haut score ("hits") dans cette première recherche BLAST.
3. La matrice PSSM remplace la matrice initiale et on effectue une 2ème recheche BLAST. 4. Les étapes 3 et 4 sont répétées et à chaque fois, les séquences nouvellement trouvées sont ajoutées afin de construire une nouvelle matrice PSSM. 5. On considère que le programme PSI-BLAST a convergé quand aucune nouvelle séquence n'est ajoutée. |
|
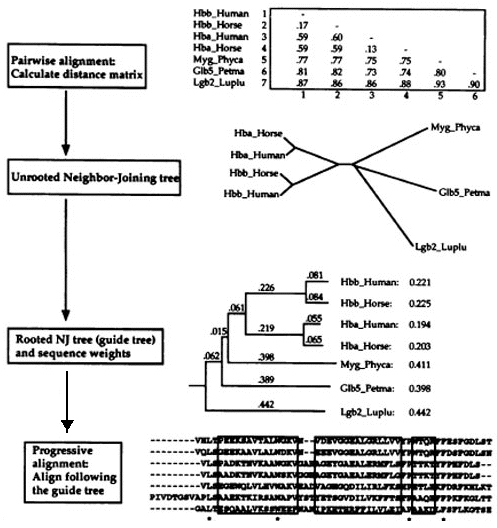
5. Programme d'alignement multiple progressif : Clustal La complexité des algorithmes de programmation dynamique croit de façon exponentielle avec le nombre de séquences à traiter, ce qui rend difficile leur utilisation pour plusieurs séquences. Pour contourner ce problème, plusieurs heuristiques ont été proposées. Le programme ClustalW utilise un algorithme d'alignement multiple progressif. Etape 1
Etape 2
Source : La Base de Connaissances en Bio-informatique Etape 3
Le risque le plus important en ce qui concerne les alignements multiples progressifs est qu'un alignement erroné à l'étape initiale engendre une erreur qui est amplifiée dans l'alignement multiple global. Le programme ClustalW comporte des particularités qui minimisent ce risque :
|
|
6. Modèle de Markov caché (Hidden Markov Model - HMM) L'apprentissage machine est un processus par lequel un ordinateur accroît ses connaissances et modifie son comportement à la suite de ses expériences et de ses actes passés. L'ordinateur apprend. Plusieurs méthodes d'apprentissage machine :
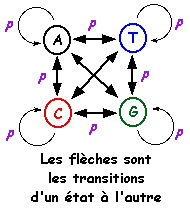
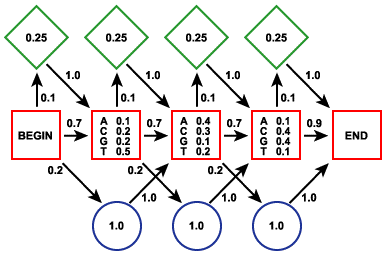
Le modèle de Markov caché est un processus stochastique. Il est apparenté aux automates probabilistes. Un tel automate est composé :
Une séquence d'événements consécutifs est donc caractérisée par le fait que la probabilité pour chaque événement de se produire ne dépend que du résultat de l'événement qui le précède. Exemple : probabilité d'observer un "U" après avoir observé "CCAAU". Le terme caché signifie que l'on observe des lettres générées par le modèle mais pas la séquence des états qui génére ces lettres. Le système nécessite une phase d'apprentissage sur un ensemble initial de séquences afin de calculer les probabilités de transitions. On applique ensuite ce système sur une nouvelle séquence et on détermine son appartenance à l'ensemble de départ. Le modèle de Markov caché ci-contre contient 3 états :
Source : La Base de Connaissances en Bio-informatique
Une probabilité est associée à chaque caractère pour l'état [appariement - mésappariement]. C'est la partie cachée de ce modèle puisque l'état dans lequel est le modèle ne définit pas la sortie (END). La base de données PFAM est un recueil de domaines complets de protéines (motifs conservés mais aussi moins conservés et insertions / délétions, figure ci-contre) obtenus à partir :
Exemples d'autres logiciels utilisant les HMM - profiles : PSI-Blast, SAM, HMMER, BLOCKS - Meta-MEME, ... |
|
7. Interprétation des résultats : E-value, P-value La signification des alignements est un point capital. Elle repose sur des valeurs spécifiques mais aussi et (peut-être surtout ?) sur une inspection visuelle du résultat par l'expérimentateur et donc sur son expertise quant aux séquences sur lesquelles il travaille. Cette signification est évaluée statistiquement en fonction de la longueur et de la composition de la séquence, de la taille de la banque et de la matrice de scores utilisée. "Sequences producing a significant alignment" : séquences ayant un alignement significatif. A chacune de ces séquences sont attribués plusieurs valeurs spécifiques qui sont une indication de la qualité de l'alignement. "High-Scoring Segment Pairs" ou "HSP" : les couples de séquences les plus longues dont les scores ne peuvent être améliorés après extension d'un segment initial (Voir une description de l'algorithme de BLAST). a. "E-Value" pour un score S (E = Expected) Pour des séquences de longueurs m et n, la statistique d'un score HSP est caractérisée par 2 paramètres de la distribution des valeurs extrêmes produites par l'algorithme de Smith-Waterman : K et λ "E-Value" est le nombre d'alignements différents que l'on peut espérer trouver dans les banques avec un score supérieur ou égal à S. C'est donc la probabilité d'observer au hasard ce score dans les banques de séquences considérées : E-Value = K.m.n. e-λS (1) "bit score S'" : ce score est dérivé du score brut S de l'alignement après normalisation. Il est utilisé pour comparer des scores provenant de recherches différentes : S' = λ.S - Ln K / Ln 2 Lien entre "E-Value" et score : E-Value = m.n. 2-S' |
|
"E-Value" |
Interprétation |
|
Plus la "E-Value" est faible, plus l'alignement est significatif. Pour des séquences requêtes trés courtes, la "E-Value" est élevée, même pour les séquences dont l'alignement obtenu est significatif. |
|
| < 1 e-100 |
La probabilité de trouver par hasard
un alignement comme celui qui est obtenu est inférieure à
1 e-100 |
| 1 e-100 < E < 1 e-50 | séquences quasiment identiques : allèles, mutations, espèces voisines |
| 1 e-50 < E < 0,1 | une éventuel lien entre la séquence requête et celles qui ont été trouvées |
| > 0,1 | séquences de l'alignement à rejeter, sans lien avec la séquences requête |
|
b. "P-Value" pour un score S Le nombre d'HSP avec un score supérieur ou égal à S et obtenus par hasard suit une distribution selon la loi de Poisson. La probabilité de ne trouver aucun HSP avec un score supérieur ou égal à S est : P = e-E E est la "E-Value" pour le score S calculée avec l'équation (1). Donc, la probabilité de trouver au moins 1 HSP avec un score supérieur ou égal à S est : P-Value = 1 - e-E |
| E | P-Value |
| 10 | 0,99995 |
| 5 | 0,993 |
| très faible | "E-Value" et de "P-Value" à peu près égales |
|
BLAST renvoie la "E-Value" plutot que la "P-Value". En effet, il est plus facile de comprendre la différence entre "E-Value" = 5 et "E-Value" = 10 qu'entre "P-Value" = 0.993 et 0.99995. |
|
| 8. Liens Internet et références bibliographiques |
|
"Cours d'autoformation en bioinformatique" - Université Paris 5 : Trés bien fait et didactique. Avec exercices corrigés d'autoévaluation. "Sequence Manipulation Suite" : ensemble d'applications Java pour manipuler les séquences. Trés bien fait et didactique pour se familiariser rapidement. "An introduction to Bionformatics Algorithms" CATH ("Class, Architecture, Topology and Homology") SCOP ("Structural Classification Of Proteins") |
|
|
Needleman, S.B. & Wunsch, C.D. (1970) "A general method applicable to the search for similarities in the amino acid sequence of two proteins" J. Mol. Biol. 48, 443 - 453 Smith, T. & Waterman M. (1981) "Identification of common molecular subsequences" J. Mol. Biol. 147, 195 - 197 |
|
|
"The Statistics of Sequence Similarity Scores" - Altschul, S.F. Altschul, S.F., Gish, W., Miller, W., Myers, E.W. & Lipman, D.J. (1990) "Basic local alignment search tool" J. Mol. Biol. 215, 403 - 410 Altschul S. F. et al. (1997) "Gapped BLAST and PSI-BLAST: a new generation of protein database search programs" Nucleic Acids Res. 25, 3389 - 3402 Naumoff D.G. & Carreras M. (2009) "PSI Protein Classifier: a new program automating PSI-BLAST search results" Molecular Biology (Engl Transl) 43, 652 - 664 |
|
|
Pearson, W.R. & Lipman, D.J. (1988) "Improved tools for biological sequence comparison" Proc. Natl. Acad. Sci. USA 85, 2444 - 244 Corpet, F. (1988) "Multiple sequence alignment with hierarchical clustering" Nucleic Acids Res. 16, 10881 - 10890 |
|
|
Thompson, J.D., Higgins, D.G. & Gibson, T.J. (1994) "CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice" Nucleic Acids Res. 22, 4673 - 4680 |
ClustalW |
| Sonnhammer et al. (1998) "Pfam: multiple sequence alignments and HMM-profiles of protein domains" Nucleic Acids Res. 26, 320 - 322 | PFAM |
|
Edgar, R.C. (2004) "MUSCLE: multiple sequence alignment with high accuracy and high throughput" Nucleic Acids Res. 32, 1792 - 1797
Biegert A. & Soding J. (2009) "Sequence context-specific profiles for homology searching" Proc Natl Acad Sci USA 106, 3770 - 3775 Thompson et al. (2011) "A Comprehensive Benchmark Study of Multiple Sequence Alignment Methods: Current Challenges and Future Perspectives" PLoS ONE 6, e18093 |
|
|
Eswaret et al. (2016) "Comparative protein structure modeling using MODELLER" Curr. Protoc. Bioinformatics Chapter 5, unit 5.6
Bramucci et al. (2012) "PyMod: sequence similarity searches, multiple sequence-structure alignments, and homology modeling within PyMOL" BMC Bioinformatics 13, S2 Braberg et al. (2012) "SALIGN: a web server for alignment of multiple protein sequences and structures" Bioinformatics 28, 2072 - 2073 |
![]()