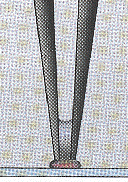| Analyse des données d'expression issues des puces à ADN |
| Tweet |
|
|
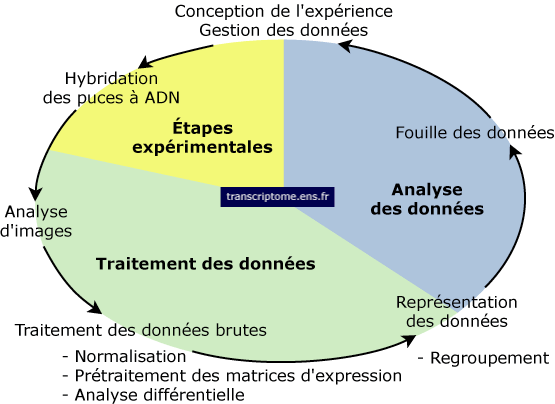
1. Introduction 2. Préparation des échantillons et hybridation 3. L'analyse des données
|
4. L'interprétation biologique des données: l'ontologie et l'annotation 5. "Chromatin ImmunoPrecipitation on Chip" 6. Comparaison des puces à ADN et de la technique de séquençage "RNA-seq" 7. Liens Internet et références bibliographiques |
|
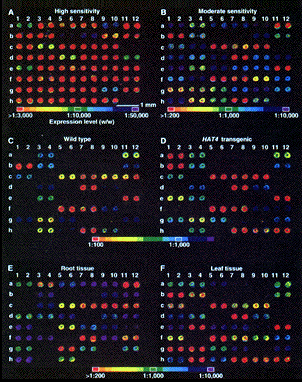
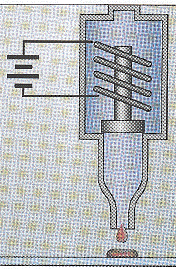
1. Introduction Préambule : les nouvelles technologies de séquençage à trés hauts débits vont-elles rendre caduques les approches telles que les puces à ADN, "Chip on Chip" ou EST ? Il y a des arguments pour (à long terme) et contre (l'acquis via les puces à ADN ou EST et la précision de cet acquis). Voir le chapitre "Comparaison puces à ADN et RNA-seq". A titre historique et conceptuel, il est malgré tout important de connaître les principes des études du transcriptome par la technique des puces à ADN. La première puce à ADN (figure ci-dessous) avec 45 sondes fluorescentes d'Arabidopsis thaliana est apparue en 1995 (Schena et al. (1995) Science 270, 467 - 470).
Le développement des puces à ADN sur membrane de nylon puis sur lame de verre a permis d'obtenir des mesures massivement parallèles de la concentration des ARN messagers d'une cellule dans un état physiologique donné. Diverses techniques permettaient à cette époque d'aborder l'étude de l'expression des gènes :
Cependant la principale avancée des puces à ADN a été de changer d'échelle : l'analyse simultanée de l'ensemble de tous les transcrits d'un génome. La technologie des puces à ADN a permis de générer des "images" de l'état de l'expression des gènes d'une cellule. L'application immédiate a été d'améliorer et de préciser le diagnostic, le pronostic et l'orientation thérapeutique dans le cas de pathologies diverses. |
| Type | "macro-array" ou filtre à haute densité | "micro-array" | puce à oligonucléotides |
| principe |
dépôt direct de l'ADN sur le support 1 condition expérimentale par puce |
dépôt direct de l'ADN sur le support 2 conditions expérimentales par puce |
sondes oligonucléotidiques synthétisées
in situ par photolithographie 1 condition expérimentale par puce |
|
marquage radioactif criblage par excès de cibles |
marquage par fluorescence | marquage par fluorescence | |
| fragments d'ADN déposés | 2400 | 10000 | jusqu'à 4,2 millions oligonucléotides |
| aperçu |
|

|

|
| Source : DNA microarray principle | |||
Principaux fabricants de puces à ADN
|
|||
|
Les puces à ADN sont des lames de verre activées sur lesquelles sont déposées de nombreuses copies d'une séquence d'ADN spécifique d'un gène donné (figure ci-dessous).
Source : Frouin & Gidrol (2005) Biofutur 252 |
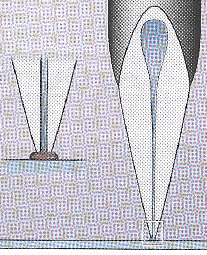
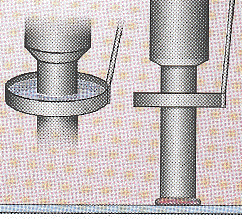
| Les différents types d'aiguilles d'impression | |
Les clavettes ou aiguilles fendues transfèrent quelques nanolitres de solution d'ADN sur l'alignement par tension capillaire quand la pointe entre en contact avec la surface. |
Les pointes et les aiguilles TeleChem appliquent de petites gouttes par contact entre l'aiguille et le support. |
La construction pointe et anneau prélève l'ADN sur un petit anneau. Une aiguille plaque la solution sur la lamelle avec une densité uniforme. |
Une imprimante à jet d'encre pulvérise des goutelettes de quelques picolitres de liquide sous pression. |
| Source des figures : "Précis de génomique" 1ère Ed. (2004) - G. Gibson & S. Muse - Ed. de Boeck Université | |
Voir une vidéo de la fabrication de puces à ADN par un robot (Université du Delaware - USA). Figure ci-contre, une description de la technologie de synthèse des oligonucléotides sur les puces ultra-haute densité.
Source : Agilent microarray technology |
|
Exemples de puces à ADN pour l'étude du transcriptome d'Arabidopsis thaliana 1. La puce CATMA ("Complete Arabidopsis Transcriptome MicroArray" - 2006) Elle contient 30 886 GSTs (étiquettes spécifiques de gènes - "Gene-specific Sequence Tags") étiquetant la majorité des gènes prédits chez Arabidopsis thaliana. Les GSTs sont des fragments génomiques de 150 à 500 paires de base amplifiés par réaction de polymérisation en chaîne (voir la position des GST). Au maxium, 50% de ces paires de base doivent être des séquences d'introns. Par ailleurs, elles ont été sélectionnées de sorte que leurs séquences ne présentent pas plus de 70% d'identité avec n'importe quelle autre séquence du génome d'Arabidopsis thaliana. La puce CATMA est complétée par 615 sondes spécifiques des génomes chloroplastique et mitochondrial. Cette puce permet :
La base de données CATdb :
2. Une puce dite "chromosomique"
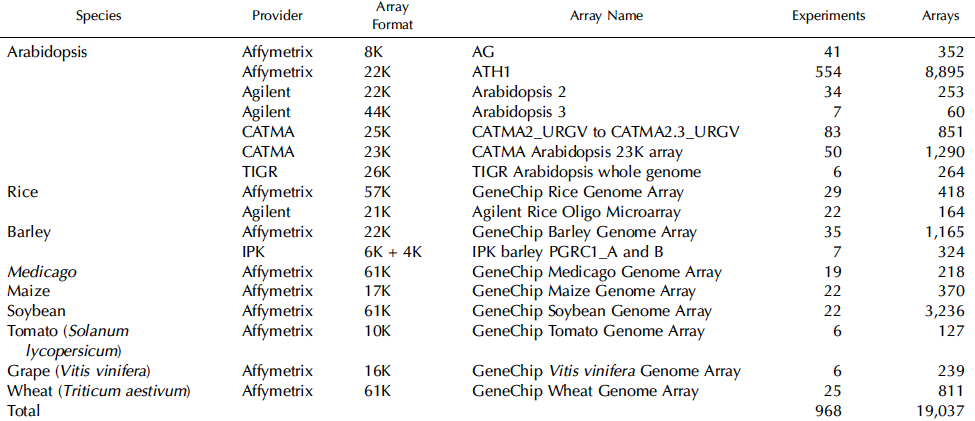
3. Une puce ATH1 d'Affymetrix Cette puce a été conçue en collaboration avec le TIGR et contient plus de 22,500 sondes oligonucléotidiques (25-mer) représentant environ 24,000 gènes d'Arabidopsis thaliana. L'interface "NetAffx Analysis Center" contient les données ATH1-12150 du TIGR et permet l'analyse des données. Voir une comparaison des caractéristiques de la puce CATMA et d'autres puces Affymetrix ("TAIR Microarray Elements Statistics"). Tableau ci-dessous : survol des puces les plus utilisées pour l'étude de la transcription des gènes chez diverses plantes et nombre d'expériences stockées dans la base de données ArrayExpress / EBI. La puce ATH1 et la puce CATMA sont les plus utilisées et plusieurs centaines d'expériences concernant Arabidopsis ont été publiées.
Source : Baginsky et al. (2010) |
|
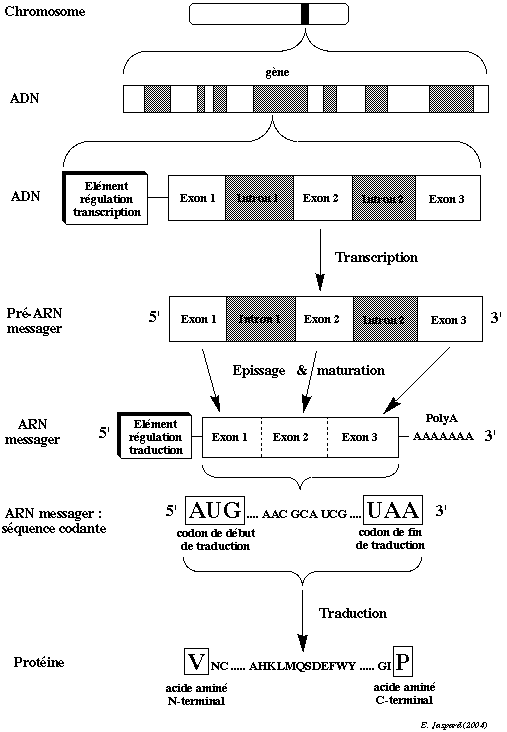
2. Préparation des échantillons et hybridation Rappel sur la transcription et la traduction Schématiquement, les deux grandes étapes de l'ADN aux ARN messagers puis des ARN messagers aux protéines sont : la transcription : synthèse de l'ARN messager à partir de l'ADN. Après la transcription, l'enchaînement des 4 nucléotides de l'ARN messager (C, G, A et U) correspond exactement à celui des 4 nucléotides (C, G, A et T) des exons de l'ADN. la traduction : synthèse de la protéine à partir de l'ARN messager. L'enchaînement des nucléotides de l'ARN messager est décodé dans les ribosomes par triplet : 3 nucléotides = 1 codon. Après la traduction, l'enchaînement des 20 acides aminés de la protéine correspond exactement à celui des codons de l'ARN messager.
|
|
Les sondes Les puces à ADN sont des lames de verre activées sur lesquelles sont déposés plusieurs milliers de "spot" d'acides nucléiques : les acides nucléiques fixés sur les puces à ADN sont appelés sondes ("probes").
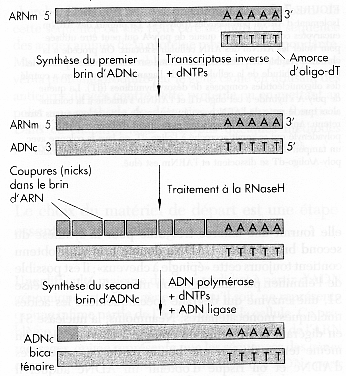
Voir une vidéo de la synthèse - dépôt des sondes par un robot. Les cibles Les acides nucléiques qui sont hybridés avec ces sondes sont appelés cibles ("targets"). Pour une exprérience donnée, une condition expérimentale (stress, pathologie, état de différenciation cellulaire, ...) est comparée à une condition de référence : les ARN messagers (les cibles) sont donc extraits des 2 types de cellules que l'on veut comparer. Les ARN messagers sont rétro-transcrits en ADNc par une transcriptase inverse (figure ci-contre). C'est une DNA polymérase qui synthétise un brin d'ADN complémentaire (ADNc) en utilisant un brin d'ARN comme matrice. Un hybride [premier brin d'ADNc - brin d'ARN] est ainsi formé dans un premier temps. Après synthèse du premier brin d'ADNc, le brin d'ARN matrice est hydrolysé par la RNAse H. Le second brin d'ADNc est ensuite synthétisé.
Source : "ADN recombinant", Watson et al. (1994) - Ed. DeBooeck Université Au cours de cette rétro-transcription :
Le marquage des cibles consiste en l'incorporation de nucléotides portant :
Ces 2 molécules sont les plus classiquement utilisées. |
| cyanine | nom | structure | longueur d'onde émission fluorescence | couleur |
| cyanine 3 (Cy3) | indodicarbocyanine 3-1-O-(2-cyanoethyl)- (N,N-diisopropyl)-phosphoramidite
|
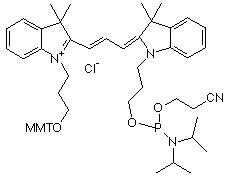 |
563 - 570 nm | vert |
| cyanine 5 (Cy5) | indodicarbocyanine 5-1-O-(2-cyanoethyl)- (N,N-diisopropyl)-phosphoramidite
|
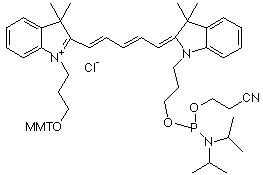 |
662 - 670 nm | rouge |
| MMT : groupe 4-monomethoxytrityle - Source : Amersham Biosciences Ltd | ||||
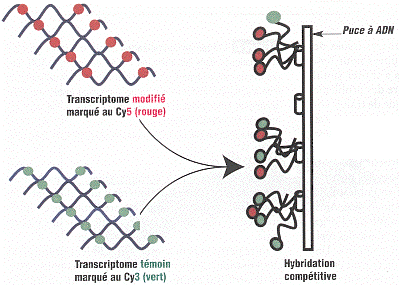
Il existe 2 méthodes de marquage des cibles en fluorescence :
Les deux familles de cibles sont mélangées et déposées sur la lame. S'il existe un brin d'ADN sonde complémentaire d'un brin d'ADNc cible, ils s'hybrident pour former de l'ADN double brin fluorescent. Cette hybridation est compétitive : plus la concentration d'un ADNc cible (donc celle de l'ARN messager qui en est l'origine) est élevée, plus l'ADNc cible s'hybridera sur la sonde.
Source : Frouin & Gidrol (2005) En conséquence, l'intensité de fluorescence traduit, respectivement :
Le rapport des intensités de fluorescence traduit donc la concentration relative des ARN messagers dans chaque condition. Ceux-ci sont soit sur-exprimés, soit exprimés de la même manière, soit sous-exprimés.
Source : Vulgariz Voir
un exemple de conditions expérimentales et de
résultats d'hybridation : "Identification of genes differentially expressed between flowers and leaves". |
|
Elle se décompose en 3 étapes :
Certains outils bioinformatiques existent pour répondre à cette démarche.
Source : "L'analyse des résultats de puces à ADN" - ENS Malgré tout, la diversité des applications des puces à ADN et des problèmatiques biologiques auxquelles elles contribuent à apporter une réponse, a nécessité le développement d'algorithmes et de logiciels spécifiques à cette technologie. |
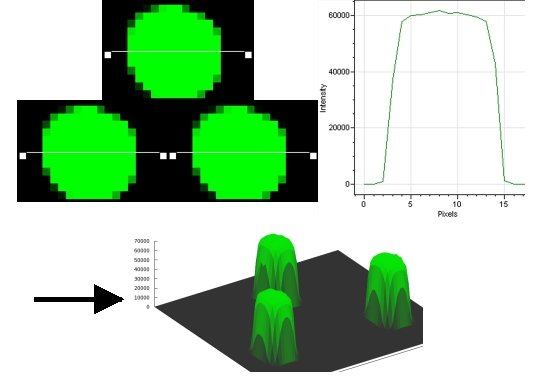
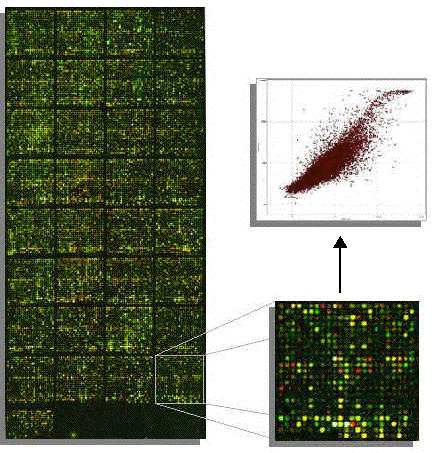
a. La détection du signal et l'analyse d'images Lors de la lecture, chaque spot est excité par un laser et l'émission de fluorescence est mesurée. On obtient 2 images en niveaux de gris qui correspondent au mélange des fluorescences respectives des 2 fluorophores. On remplace les niveaux de gris par :
Après superposition, on obtient une image en fausses couleurs composée de spots :
Source : Frouin, V. & Gidrol, X. (2005) Ces étapes font appel à des techniques de traitement de l'image et utilisent des algorithmes de morphologie mathématique. Les technologies pour l'analyse des images sont de plus en plus performantes. La résolution est augmentée, en conséquence :
Par ailleurs, de nouvelles surfaces sont utilisées pour remplacer le verre. Par exemple, des cristaux de mélange d'oxyde de silice et de titane à fluorescence accrue.
Source : Agilent microarray technology
Source : MYcroarray |
|
b. Le traitement des données brutes Après l'étape d'analyse de l'image, chaque sonde est caractérisée par :
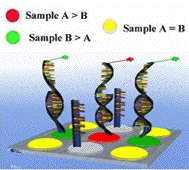
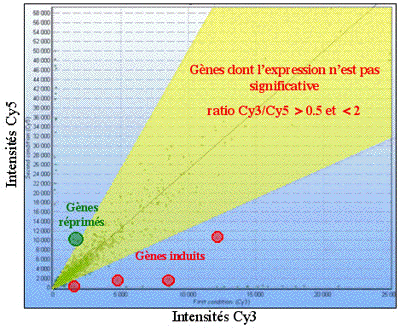
Les signaux rouges et vert ne peuvent être interprétés séparément. Les puces à ADN permettent de mesurer une variation de transcription d'un gène entre 2 conditions expérimentales (référence et pathologique, par exemple). Elles fournissent donc des valeurs relatives. Pour chaque spot, le logarithme du rapport (r) de l'intensité de fluorescence de la condition pathologique sur l'intensité de fluorescence de la condition de référence est calculé (rapport fluorescence rouge / fluorescence verte) : log2(r). Ce rapport permet d'évaluer la différence du taux de transcription d'un gène entre les 2 échantillons biologiques étudiés. On considère pour un rapport :
Source : Le principe des puces à ADN (Cours ENS) Voir un exemple : Puce CATMA / Cy3 - feuilles / Cy5 : fleurs / Arabidopsis thaliana |
|
Normalisation des données de fluorescence Ci-dessous : exemple de puces de 16000 oligonucléotides de Medicago truncatula et une représentation des intensités des spots.
Source : The Samuel Roberts Noble Foundation Elle a pour but, entre autre, de distinguer les variations aléatoires (biologiques et expérimentales : celles que l'on veut mettre en évidence) des variations systématiques. Ces dernières ont pour origine en particulier :
L'hypothèse de base de la normalisation est que la majorité des gènes ont un niveau d'expression invariant entre 2 conditions (référence et pathologique, par exemple), soit : log2(r) = 0. La normalisation a donc pour but de ramener la moyenne de cette grandeur à 0. Exemple de valeurs normalisées : le rapport permet de mettre en évidence les gènes pour lesquels le canal rouge (condition pathologique) donne une valeur supérieure au canal vert. Le log2(r) donne une distribution symétrique autour de zéro. Enfin, la soustraction du rapport moyen des logarithmes permet de tenir compte de l'intensité plus importante du canal rouge. |
| intensité Rouge | intensité Vert | Différence | Rapport ( V/R) | log2(r) | Centrage de R |
| 16500 | 15104 | -1396 | 0,915 | -0,128 | -0,048 |
| 357 | 158 | -199 | 0,443 | -1,175 | -1,095 |
| 8250 | 8025 | -225 | 0,973 | -0,039 | 0,040 |
Le filtrage Un rapport d'une valeur donnée peut être
obtenu par des valeurs d'intensité [rouge/vert] trés proches
du bruit de fond (peu fiables alors) ou, au contraire, trés élevées
(plus significatives). Le filtrage a pour but d'éliminer les sondes pour lesquelles une des mesures d'intensité de fluorescence est inférieure à un seuil (arbitraire ou déterminé à partir d'un modèle). Risque statistique Les traitements précédents aboutissent à une liste de rapports (r) pour chaque gène. La suite consiste à déterminer, à l'aide de logiciels utilisant des techniques statistiques, les gènes différentiellement exprimés (ceux dont les valeurs de log2(r) sont significativement différents de 0). Cependant, le choix d'une méthode d'analyse est liée aux conditions dans lesquelles a été menée l'expérience (réplicats, facteurs expérimentaux, ...). De plus, ces outils informatiques ne donnent pas de valeur seuil sur le résultat d'un test pour évaluer si l'expression d'un gène est modulée ou non. Il incombe à l'expérimentateur de choisir son niveau de risque. La standardisation La méthode de standardisation "MIAME" ("Minimum information about a microarray experiment") est une charte qui décrit l'information minimale (à propos d'une expérience de puce à ADN) requise pour que les résultats de cette expérience soient interprétables, d'une manière non-ambigüe et de sorte que cette expérience soit reproductible. Tout expérimentateur qui désire déposer ces données issues de puces à ADN dans une banque doit répondre à cette charte en indiquant (entre autre) :
|
|
c. Analyse des données : la prédiction Outre l'obtention de listes de gènes différentiellement transcrits, on peut suivre le profil de transcription d'un gène : l'ensemble des valeurs de transcription mesurées dans des conditions diverses ou au cours d'une étude cinétique. Dans ce cas, l'une des 2 sources d'ARN hybridés est fixée de sorte que toutes les valeurs de log2(r) soient comparables. Cette source est alors considérée comme la référence. On peut dés lors s'intéresser :
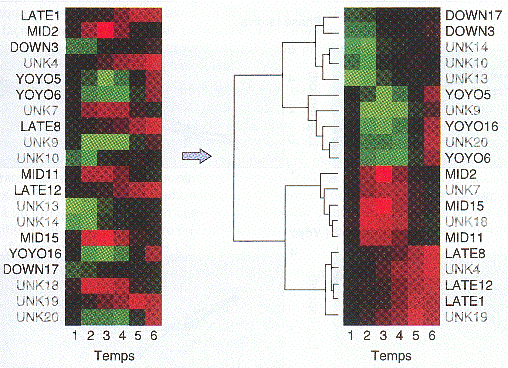
Exemples de logiciels de regroupement : "J-Express" - "MultiExperiment Viewer" - "Genesis" Figures ci-dessous : une série de profils d'expression de gènes désordonnés (figure de gauche) peut être convertie en une série de groupes par le regroupement hiérachique (Eisen et al., 1998).
Le résultat (à droite) est un arbre qui montre l'évolution de l'expression dans le temps pour certains gènes hypothétiques.
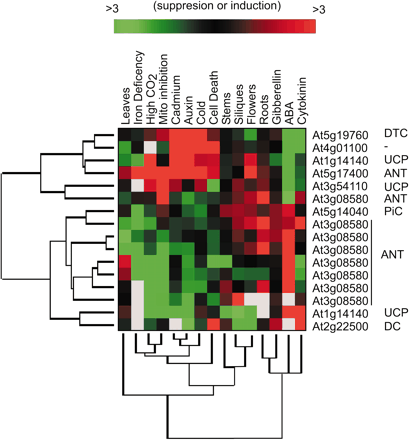
Source : "Précis de génomique" (2004) - G. Gibson & S. Muse Figure ci-dessous : différence de profils d'expression des gènes mitochondriaux de la famille des transporteurs de Arabidopsis thaliana. Les résultats mettent en évidence les variations selon type de tissus et la réponse à des stress hormonaux et environnementaux.
Source : Millar & Heazlewood (2003) "Genomic and Proteomic Analysis of Mitochondrial Carrier Proteins in Arabidopsis" Plant Physiol. 131, 443 - 453 |
|
4. L'interprétation biologique des données : l'ontologie et l'annotation L'interprétation biologiques des données issues des puces à ADN (et d'autres technologies) nécessite de corréler les résultats de ces données à des informations encyclopédiques contenues dans certaines bases de données.
Source : CBB group (Berlin) a. L'ontologie Une ontologie est un ensemble structuré de termes et de concepts qui représentent le sens d'un champ d'informations, que ce soit par :
Chaque terme de l'ontologie est associé à des "lexicons" (synonymes, homonymes, hyperonymes, ...). Le réseau autour d'un terme est appelé concept. Les concepts sont formalisés sous forme d'un graphe au sein duquel il existe des relations sémantiques ou d'inclusion ("appartient à"). |
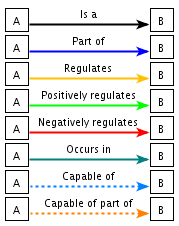
| "is-a" ("est un") |
symbole |
 |
| "part-of" ("fait partie de") | symbole |
b. Le consortium Gene Ontology De manière schématique, on peut considérer qu'en génomique, l'ontologie est associée aux notions de terminologie et de classification. Le consortium Gene Ontology (GO) :
|
|
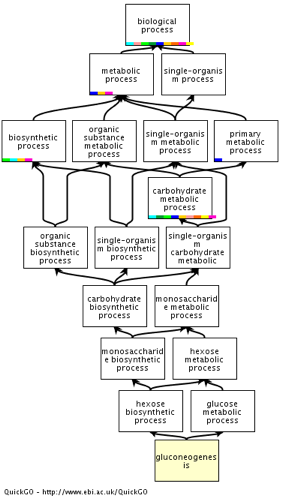
Exemple : tous les termes suivants
|
décrivent la formation du glucose (néoglucogénèse) |
D'où le terme GO : "gluconeogenesis" dont l'identifiant est : GO:0006094 |
Le produit d'un gène :
GO décrit donc les produits des gènes via un ensemble de termes au sein d'un graphe dirigé acyclique ("Directed Acyclic Graph" - DAG) qui contient 3 axes hiérarchiquement indépendants (CC, BP et MF). Les termes de GO (les noeuds de l'ontologie) sont liés par un ensemble de relations. En particulier : "Is a", "Part of", "Regulates", "Positively Regulates" et "Negatively Regulates".
Chaque terme hérite de la signification de tous les termes qui le séparent de la racine de l'ontologie (notion d'ancêtre, parent et enfant).
Le niveau de preuve d'une annotation est précisée par des codes ("Evidence Codes") répartis en catégories : "Experimental", "Computational analysis", "Author statement" (déclaration d'auteur), "Curatorial statement" (déclaration de curateur), "Automatically-assigned". La dernière catégorie est "Automatically-assigned" (annotation automatique) dont le sous-code "Inferred from Electronic Annotation" (IEA) représente environ 95% de l'annotation. Voir "Evidence Code Decision Tree". |
| Exemple de code | information déduite |
| IMP | du phénotype des mutants |
| IGI | d'interactions génétiques |
| IPI | d'interactions physiques |
| ISS | par analogie de séquences ou de structure |
| IDA | par expérimentation directe |
| IEP | du profil d'expression |
| IEA | par l'annotation in silico |
| TAS | à partir de la publication de résultats fiables |
| NAS | à partir de la publication de résultats non vérifiables |
Enfin, on ne soulignera jamais assez le le rôle primordial des scientifiques que l'on nomme curateurs. Ils effectuent, grâce à leur immense culture, un travail dans l'ombre qui assure la qualité, la rigueur et la pertinence des informations associées aux données de génomique, transcriptomique, protéomique et autres contenues dans les bases de données. GO est extrêmement complexe et nécessite un "navigateur" dans l'arbre de l'ontologie. Le plus utilisé pour GO est AmiGO2. |
c. Exemples de logiciels et d'interfaces web pour l'annotation 1. Le consortium GO propose un ensemble de logiciels ("Gene Ontology Tools") pour traiter et analyser des données de divers types, en particulier celles issues des puces à ADN. Ces logiciels sont utilisables directement via une interface Web ou à installer sur l'ordinateur pour divers types de systèmes d'exploitation (Unix, Linux, Windows, Mac) 2. L'une des interfaces les plus didactiques et intuitives pour l'annotation : "QuickGO (GO Browser)". 3. Autre exemple de logiciel - interface web : "GOrilla". Un exemple trés didactique de classification hiérarchique et d'ontologie est montré avec le lien "Running example". 4. Voir un cours sur l'annotation. d. Exemples de bases de données de niveau de transcription de gènes
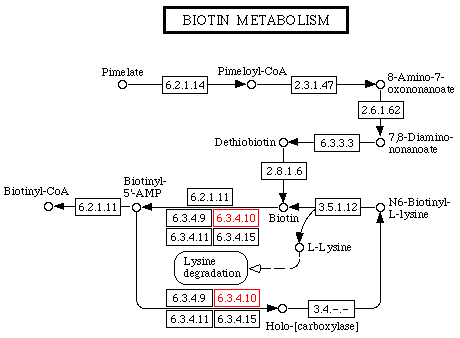
e. La collection de bases de données KEGG Il propose pour les voies métaboliques et les métabolites impliqués dans ces voies, des graphes d'interaction entre les enzymes impliquées dans ces voies et, par extension, entre les gènes qui codent ces enzymes.
Source : KEGG C'est un outil puissant pour la métabolomique. Exemple : la biotine existe sous forme libre ou sous forme de groupement prosthétique lié à certaines carboxylases qui catalysent des réactions de synthèse des acides gras ou de certains acides aminés. En allant sur le site, l'image originale est interactive. En cliquant sur les N° EC ou les noms, on accéde à une multitude d'informations sur les molécules choisies. |
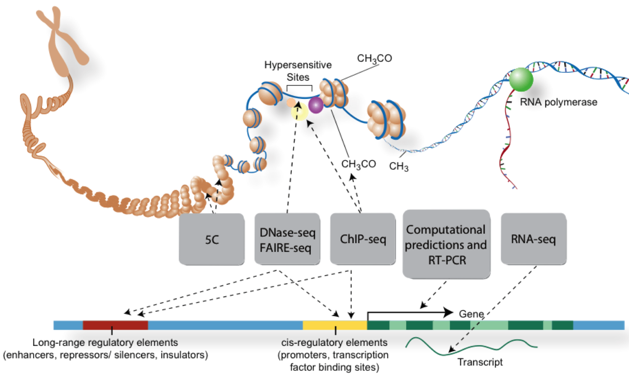
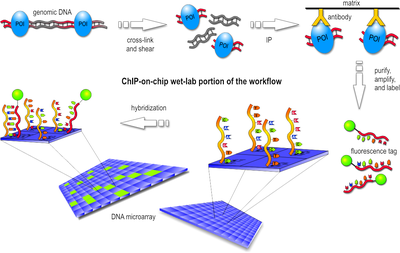
5. "Chromatin ImmunoPrecipitation on Chip" Cette méthode appelée aussi "ChIP on chip" permet d'identifier les protéines qui se fixent à l'ADN. Elle est extrêmement utile pour l'étude des sites de fixation des facteurs de transcription, ou les histones (étude des profils épigénétiques), par exemple. Figure ci-contre : Techniques de traitement des acides nucléiques avant séquençage pour l'analyse de parties spécifiques des génomes.
Source : ENCODE Par exemple :
La méthode "ChIP on chip" combine celle de l'immunoprécipitation de la chromatine et celle des puces à ADN. On crée d'abord une liaison covalente in vivo entre les protéines et la partie de l'ADN avec lesquelles elles interagissent. On utilise la formaldéhyde en général.
Source : Wikipédia L'ADN de la cellule est extrait puis découpé en courts fragments. On sélectionne les fragments d'ADN qui sont associés à la protéine étudiée avec un anticorps spécifique de cette protéine. Les complexes [ADN-protéine-anticorps] sont précipités. Cette précipitation élimine l'ADN qui ne s'est pas associé à la protéine étudiée. La partie protéique du complexe [ADN-protéine-anticorps] est protéolysé afin de ne conserver que l'ADN. En conséquence, les courts fragments d'ADN récupérés sont ceux qui interagissent avec la protéine étudiée. Ces fragments sont identifiés par la technique des puces à ADN. |
| Eléments du génome cartographiées | Techniques utilisées |
| Régions transcrites en ARN | RNA-seq / CAGE / RNA-PET |
| Régions codant des protéines | Spectromètrie de masse |
| Sites de fixation des facteurs de transcription | ChIP-seq / DNase-seq |
| Structure de la chromatine | DNase-seq / FAIRE-seq / Histone ChIP-seq / MNase-seq |
| Sites de méthylation de l'ADN | RRBS |
|
Définitions des acronymes des nouvelles technologies
ChIPBase : base de données et plate-forme pour le décodage des cartes de liaison, des facteurs de transcription, des profils d'expression, de la régulation de la transcription de longs ARN non codants ("long non-coding RNAs" : lncRNAs, lincRNAs), de microRNA et autres ARN non codant (snoRNAs, tRNAs, snRNAs, ...) et des gènes codant des protéines. |
|
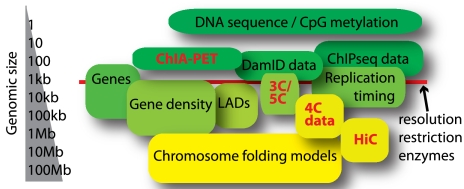
*Le "chromosome interactome" : le super-enroulement et compacité de l'ADN dans les chromosomes (chromatine, histone) : une fois déplié, la molécule d'ADN d'une cellule de l'homme mesure environ 2 m, soit 200.000 fois le diamètre moyen du noyau d'une cellule de mammifère.
Source : de Wit & de Laat (2012) L'exploration du "chromosome interactome" et des interactions chromatine-chromatine à longue distance in vivo est liée au développement de nouvelles technologies incluant le séquençage à très haut débit :
|
6. Comparaison des puces à ADN et de la technique de séquençage "RNA-seq" Voir un développement concernant la technologie RNA-seq. a. Les puces à ADN et la technique RNA-seq ont toutes deux une haute reproductibilité de résultats avec des réplicats biologiques. b. Les puces à ADN permettent difficilement de distinguer le cas "pas de transcription" du cas "très faible transcription". c. En raison de la différence de transcription des gènes et/ou du nombre de gènes codant un même type d'ARN messagers, il n'existe dans une cellule que quelques copies de certains ARN messagers et des dizaines de milliers de copies d'autres ARN messagers :
d. De multiples transcrits sont générés à partir de certains gènes par épissage alternatif. L'un des avantages de la technique RNA-Seq est sa capacité à détecter ces isoformes différentiellement transcrites :
e. Les puces à ADN sont sujettes à une saturation d'hybridation en ce qui concerne les transcrits très abondants. Elles ne peuvent pas fournir des mesures quantitatives fiables des changements subtils de la transcription de gènes abondants. f. La technique RNA-Seq permet d'identifier des variants d'un seul nucléotide ("single nucleotide polymorphism" - SNP). La technique RNA-Seq présente deux avantages dans la détection de variants génétiques :
g. La technique RNA-Seq permet :
Pour l'instant la technique RNA-seq présente deux inconvénients :
Par ailleurs, l'un des atouts actuels (mais qui ne peut que diminuer avec le temps) des puces à ADN est l'acquis des dizaines de milliers d'expériences qui ont été menées avec cette technique et les différentes annotations des transcriptomes issues de toutes ces expériences. L'un des atouts de la technique RNA-seq (ou d'une autre technologie à venir) est l'évolution très rapide des technologies de séquençage à très haut débit : le développement des méthodes avec multiplexage par répartition codes barres, des lectures ("reads") plus longues et un plus grand nombre de lectures appariées ("paired end reads"). Pour l'instant les puces à ADN et la technique RNA-seq restent donc complémentaires et peuvent même être combinées avec des résultats très importants. |
| 7. Liens Internet et références bibliographiques |
|
Consortium "Gene Ontology" (GO) KEGG ("Kyoto Encyclopedia of genes ans genomes") GEO ("Gene Expression Omnibus") TAIR |
|
|
Barrett et al. (2005) "NCBI GEO : mining millions of expression profilesÐdatabase and tools" Nuc. Acids Res. 33, D562 - D566 Frouin, V. & Gidrol, X. (2005) "Analyse des doinnées d'expression issues des puces à ADN" Biofutur 252, 22 - 26 Schena et al. (1995) "Quantitative monitoring of gene expression patterns with a complementary DNA microarray" Science 270, 467 - 470 Schena et al. (1996) "Parallel human genome analysis: Microarray-based expression monitoring of 1000 genes" PNAS 93, 10614 - 10619 Lockhart et al. (1996) "Expression monitoring by hybridization to high-density oligonucleotide arrays" Nat. Biotechnol. 14, 1675 - 1680 DeRisi et al. (1997) "Exploring the metabolic and genetic control of gene expression on a genomic scale" Science 278, 680 - 686 Eisen et al. (1998) "Cluster analysis and display of genome-wide expression patterns" PNAS 95, 14863 - 14868 Lipshutz et al. (1999) "High density synthetic oligonucleotide arrays" Nat. Genetics 21, 20 - 24 Alizadeh et al. (2000 ) "Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling" Nature 403, 503 - 511 Garcia-Hernandez et al. (2002) "TAIR: a resource for integrated Arabidopsis data" Functional & Integrative Genomics 2, 239 - 253 |
|
|
Wang et al. (2007) "A new method to measure the semantic similarity of GO terms" Bioinformatics 23, 1274 - 1281 Zhidian et al. (2009) "G-SESAME: web tools for GO-term-based gene similarity analysis and knowledge discovery" Nuc. Acids. Res. 37, W345 - W349 de Wit & de Laat (2012) "A decade of 3C technologies: insights into nuclear organization" Genes Dev. 26, 11 - 24 Zhao et al. (2014) "Comparison of RNA-Seq and Microarray in Transcriptome Profiling of Activated T Cells" PLoS ONE 9, e78644 |
|
![]()