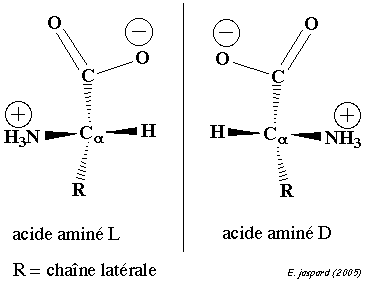
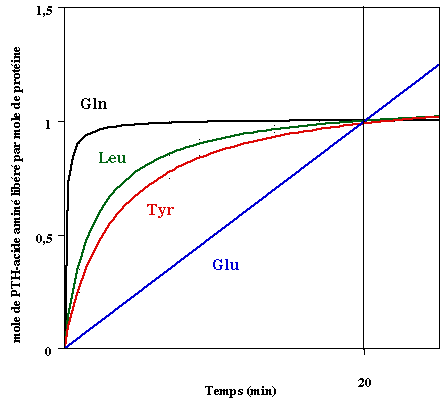
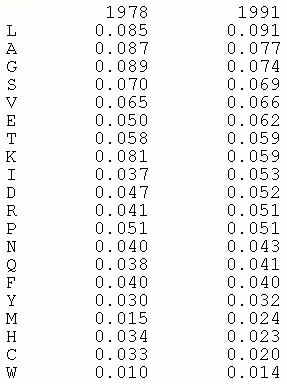
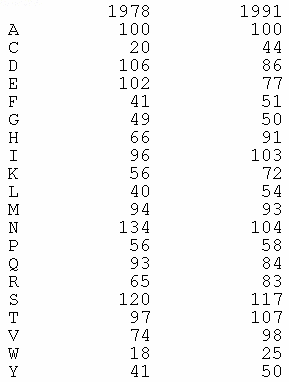
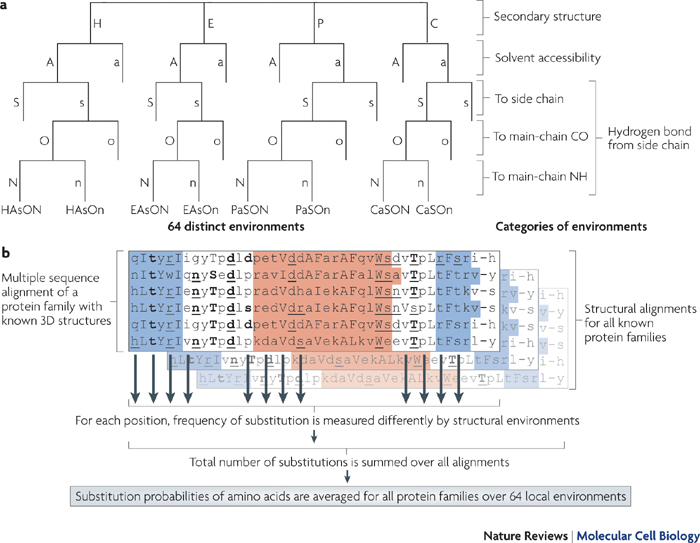
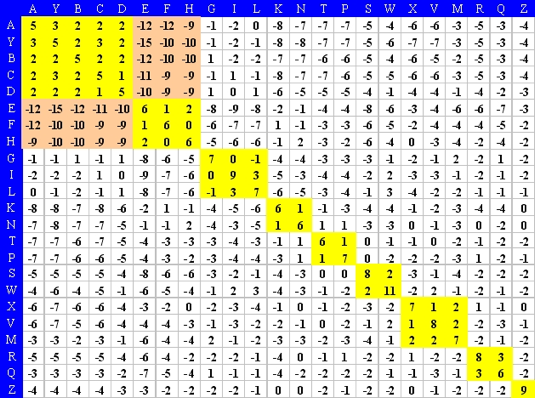
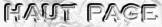Les compositions en acides aminés de 4 jeux de données "protéines désordonnées" (dis XRAY, dis NMR, dis CD, dis Fam32) ont été comparées entre elles et avec celle d'un jeu de données "protéines ordonnées".
La proportion de chaque acide aminé dans chacun des jeux de données a été exprimée par le rapport : [(nombre de l'acide aminé considéré dans les protéines désordonnées) - (nombre de l'acide aminé considéré dans les protéines ordonnées)] / (nombre de l'acide aminé considéré dans les protéines ordonnées).
Dans la figure ci-dessous, un pic négatif signifie donc que le jeux de données "protéines désordonnées" considéré contient moins l'acide aminé considéré que le jeu de données "protéines ordonnées".
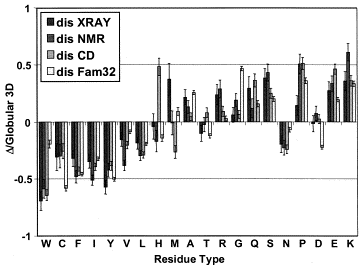
Source : Dunker et al. (2001)
Les acides aminés sont rangés en fonction de leur indice de fléxibilité corrigé par le facteur de température ("Debye-Waller factor" ou "B-factor") qui tient compte des mouvements dûs à la châleur sur l'atténuation de la diffraction des rayons X. Celà permet de tenir davantage compte de certains effets de l'environnement sur les acides aminés.
L'acide aminé le moins flexible est à gauche (Trp) et le plus flexible est à droite (Lys).
Si on représente la valeur absolue de la charge nette moyenne (c'est-à-dire pondérée par la longueur de la chaîne polypeptidique de l'IDP considérée) à pH 7 (<R>) en fonction de la valeur absolue de l'hydrophobicité moyenne (<H>), on obtient un graphique avec deux zones qui correspondent aux IDP et aux protéines structurées, respectivement.
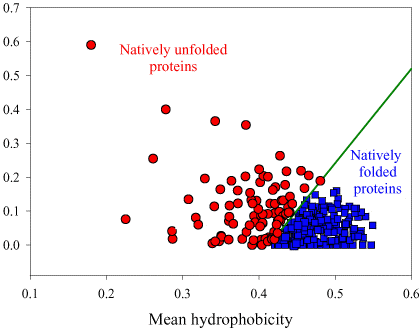
Source : Uversky et al. (2000)
Ces zones sont délimitées par une droite d'équation : <H> = [ <R> + 1,151 ] / 2,785 et les IDP sont au dessus de cette ligne.
On obtient un graphique équivalent si on représente <R> en fonction de la valeur absolue de l'hydropathie moyenne ("GRand Average of hYdropathy" - <GRAVY>). |