| Programmation dynamique et phylogénie (globines, insuline et peroxydases de classe III) |
Voir les documents de cours et les documents pour le TD sur l'espace Moodle. |
|
Partie I : algorithme de Needleman & Wunsch
|
Principe a. On construit un tableau ou matrice de la manière suivante : |
|
Lignes i
|
Colonnes j
|
La case de "départ" (en haut à gauche de la matrice) est la case (i = 0, j = 0). La case "d'arrivée" ("en bas à droite de la matrice) est la case (i = n, j = m). |
|
| ligne i / colonne j | - (gap) | A | T |
| - (gap) | score (i = 0, j = 0) | ||
| A | score (i = 1, j = 1) | score (i = 1, j = 2) | |
| G | score (i = 2, j = 1) | score (i = 2, j = 2) | |
| C | score (i = 3, j = 1) | score (i = 3, j = 2) |
b. Puis on calcule le score (i, j) de chacune des cases, ligne par ligne, selon 3 opérations possibles : insertion, délétion ou correspondance qui peut-être une identité (Xi, Xj) ou une substitution (Xi, Yj). Ce score est calculé de 3 façons qui traduisent les 3 "déplacements" possibles d'une case à l'autre :
c. Le score (i, j) de chacune des cases de cette matrice est la valeur maximale des " déplacements possibles (↘), (➞) ou (↓). |
|
Application : comparer les 2 séquences suivantes : séquence 1 : ATTCAAGCTGA Avec les 3 paramètres de scores : identité = 4, substitution = -1 et insertion/délétion ("gap") = -2 (pénalité). |
a. Matrice des coûts |
| i/j | - | A | A | C | T | T | G | C | G | T | G | A |
| - | 0 | -2 | -4 | -6 | -8 | -10 | -12 | -14 | -16 | -18 | -20 | -22 |
| A | -2 | 2 | 0 | |||||||||
| T | -4 | 8 ➞ | c = 6 | |||||||||
| T | -6 | |||||||||||
| C | -8 | -2 ↘ | -1 | |||||||||
| A | -10 | -4 | a = 2 | 3 ↘ | 4 | |||||||
| A | -12 | 1 | b = 2 | |||||||||
| G | -14 | |||||||||||
| C | -16 | 11 ↘ | 9 | |||||||||
| T | -18 | 9 | d = 10 | |||||||||
| G | -20 | 17 ↘ | 15 | |||||||||
| A | -22 | 15 | e = 21 |
Exemple pour la case a (i = 5, j = 2) :
Exemple pour la case c (i = 2, j = 6) :
|
b. Matrice des chemins
Exemple pour la case a' : déplacement suivi en diagonale donc le score de la case a' = 0. |
| - | A | A | C | T | T | G | C | G | T | G | A | |
| - | 0 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| A | -1 | |||||||||||
| T | -1 | horizontal c' = 1 |
||||||||||
| T | -1 | |||||||||||
| C | -1 | |||||||||||
| A | -1 | diagonale a' = 0 |
||||||||||
| A | -1 | diagonale b' = 0 |
||||||||||
| G | -1 | |||||||||||
| C | -1 | |||||||||||
| T | -1 | diagonale d' = 0 |
||||||||||
| G | -1 | |||||||||||
| A | -1 | diagonale e' = 0 |
c. Alignement de seq1 et seq2 On part de la case (i = n, j = m) en bas à droite de la matrice des chemins (dernière position de l'alignement A versus A) et on remonte (↖ ou ← ou ↑) le chemin parcouru. |
| - | A | A | C | T | T | G | C | G | T | G | A | |
| A | 0 | |||||||||||
| T | ↑ -1 | |||||||||||
| T | ↖ 0 | |||||||||||
| C | ↖ 0 | |||||||||||
| A | ↖ 0 | |||||||||||
| A | ↖ 0 | |||||||||||
| G | ↖ 0 | |||||||||||
| C | ↖ 0 | ← 1 | ||||||||||
| T | ↖ 0 | |||||||||||
| G | ↖ 0 | |||||||||||
| A | ↖ e'= 0 |
| On obtient l'alignement suivant (paramètres de scores : identité = 4, substitution = -1, indel = -2) : | ||||||||||||
| seq2 | A | - | A | C | T | T | G | C | G | T | G | A |
| seq1 | A | T | T | C | A | A | G | C | - | T | G | A |
| S | 4 | -2 | -1 | 4 | -1 | -1 | 4 | 4 | -2 | 4 | 4 | 4 |
Le score global de l'alignement = 21. |
||||||||||||
Partie II : PHYLOGENIE La phylogénie étudie les liens de parenté (donc l’évolution et la classification) entre organismes Le clade est l’unité élémentaire de la classification phylogénétique.
Définitions des termes : clade, cladogramme, parcimonie, pas évolutif, synapomorphie, autapomorphie, symplésiomorphie, homoplasie, réversion, diagramme de Venn … => voir l'article Wikipédia "cladistique". |
| La phylogénie permet de différencier des gènes homologues descendant d'un même gène ancestral. | |
Duplication
|
Spéciation
|
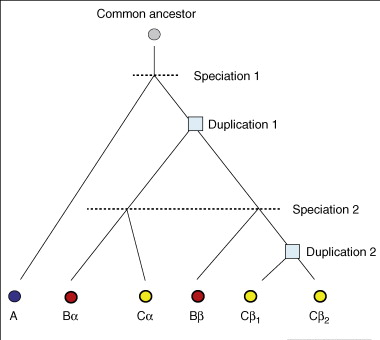
L'arbre ci-contre décrit l'évolution de 6 gènes (cercles) homologues de 3 espèces A, B et C (couleurs) :
Source : Studer & Robinson-Rechavi (2009) |
|
La duplication des gènes est le principal mécanisme générateur de nouveaux gènes et de nouveaux processus biologiques qui a facilité l’évolution des organismes simples vers les organismes complexes. Devenir des gènes après duplication :
|
Exemples de méthodes pour générer un arbre phylogénétique La méthode UPGMA ("Unweighted Pair Group Method with Arithmetic Mean") construit un arbre enraciné sur la base d'une matrice de distances entre toutes les paires de séquences analysées. La méthode NJ ("Neighbour Joining") qui recherche de manière séquentielle des voisins en minimisant la longueur totale de l'arbre. Elle génère des arbres non enracinés. Elle est rapide et adaptée à des arbres de plusieurs milliers de séquences. La méthode ML ("Maximum Likelihood") est basée sur un ou plusieurs caractère(s) à étudier. Elle nécessite un modèle d'évolution probabiliste (dont le choix est déterminant) qui permet d'évaluer, en termes de probabilités (vraisemblance), l'ordre des embranchements et la longueur des branches de l'arbre. La méthode MP ("Maximum Parcimony") recherche parmi les arbres possibles et les séquences possibles de noeuds ancestraux, la combinaison qui nécessite le plus petit nombre de changements évolutifs. Approche Bayesienne (exemple : logiciel "MrBayes") : L'inférence bayésienne de la phylogénie ("Bayesian phylogenetic tree") est une méthode pour estimer la probabilité qu'un arbre soit correct sur la base des données analysées, de "l'à priori" et du modèle de vraisemblance employés pour construire cet arbre. Théorème de Bayes : il permet de déterminer la probabilité d'un évènement compte-tenu des informations connues et de données nouvelles. Il s'écrit : P(A|B) = [P(B|A) . P(A)] / P(B)
|
La longueur des branches
Arbre enraciné ou arbre non-enraciné Plusieurs méthodes pour tenter d'enraciner un arbre phylogénique :
Valeur de "bootstrap" Pour compléter la construction d'un arbre phylogénétique, sa robustesse doit être évaluée, en général par une valeur de "bootstrap" (de 0 à 100%) associée à chaque branche de l'arbre :
|
Exercice 1 : généralités sur les globines a. Structure des globines Le repliement de type "globine" est un repliement à 8 hélices α trouvé chez les bactéries et les eucaryotes et donc conservé au cours de l'évolution.
b. Classification des globines Elles ont évolué à partir d'un ancêtre commun et peuvent être divisées en 3 groupes : les globines à domaine unique et 2 types de globines dites "chiméres" (les flavo-hémoglobines et les capteurs couplés à la globine - "globin-coupled sensors"). Principaux types de globines :
|
Exercice 1 : étude de la famille des gènes de l'hémoglobine humaine Les sous-unités de l'hémoglobine (Hb) sont codées par des gènes de la famille des globines α et β. Les ancêtres de ces gènes sont apparus après une duplication il y a plusieurs centaines de millions d'années.
|
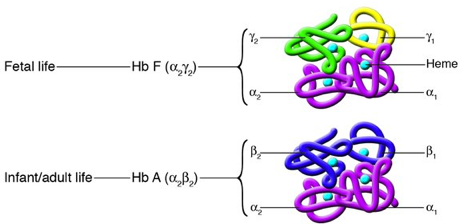
Types d'hémoglobine Il existe plusieurs formes d'hémoglobine qui sont toutes des hétérotétramères :
|
|
| Fonction de l'hémoglobine | Elle est impliquée dans le transport du dioxygène. L'hémoglobine est une hémoprotéine : chaque chaîne polypeptidique contient un groupe hème constitué d'un noyau porphyrine fixant un atome de fer. |
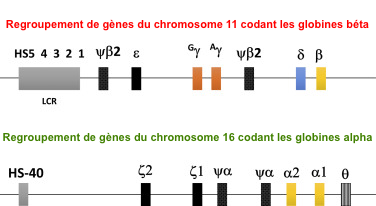
| Les gènes codant les globines chez l'homme forment une famille multigénique. |
|
Gènes codant chaque chaîne polypeptidique Les gènes sont regroupés ("cluster").
|
|
Il existe :
dont les séquences sont très similaires de celles des gènes fonctionnels correspondants. Cependant, ils contiennent des codons de terminaison et d'autres mutations qui empêchent la synthèse d'ARN messager codant des hémoglobines fonctionnelles. |
|
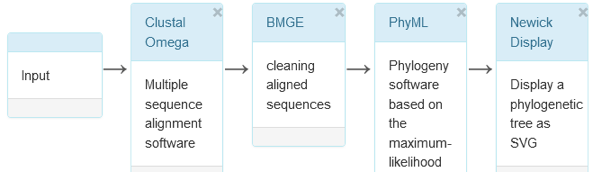
Question 1 : construction et visualisation de l'arbre avec les séquences des globines La figure ci-dessous décrit l'enchaînement des programmes "PhyML - OneClick" de la suite logicielle NGPhylogeny pour analyser les séquences FASTA des hémoglobines de l'homme et obtenir un arbre phylogénétique.
Source : NGPhylogeny |
| Récupération des séquences des globines humaines au format FASTA. | Aller à Uniprot.
Résultat : Séquences FASTA des 9 globines de l'homme. |
| Le programme MAFFT ("Multiple Alignment using Fast Fourier Transform" - alignement multiple utilisant la transformée de Fourier rapide) | |
Ce programme génère plusieurs alignements de séquences (acides aminés ou nucléotides) multiples. A l'origine, l'algorithme de MAFFT effectuait un alignement progressif, les séquences étant regroupées par transformée de Fourier rapide.
|
L'algorithme MAFFT suit 5 étapes :
|
Le programme BMGE ("Block Mapping and Gathering with Entropy") : cartographie et collecte de blocs avec entropie. |
Ce programme sélectionne, au sein d'un alignement de séquences multiples (nucléotides ou acides aminés), des régions adaptées à l'inférence phylogénétique.
|
Le programme PhyML utilise la méthode statistique du maximum de vraisemblance et fait appel à des heuristiques.
|
Le format Newick (extension : .nwk) C'est un format de fichier bioinformatique de données de relations phylogénétiques pour représenter un arbre :
Exemple de résultat avec les globines : (sp_P09105_HBAT_HUMAN_Hemoglobin_subunit_theta_1,sp_P69905_HBA_HUMAN_Hemoglobin_subunit_alpha, Voir une description du format Newick. |
L'outil iTOL ("Interactive Tree Of Life") C'est un outil en ligne (exécuté sur le serveur de l'EMBL) pour afficher, annotater, modifier et gérer des arbres phylogénétiques avec différents jeux de données. |
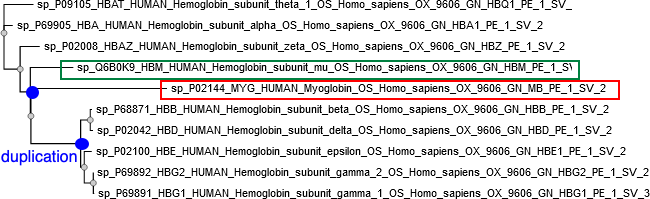
Question 1 : quelques commentaires des résultats MSA : "Multiple Sequence Alignment".
|
On voit bien la duplication qui a engendré les chaînes alpha et la duplication qui a engendré les chaînes béta.
|
Source : NGPhylogeny |
Sur la figure ci-dessus, il est difficile d'estimer le nombre d'évènements de duplication car la résolution n'est pas suffisante. Tous les noeuds correspondent à des duplications car toutes les séquences sont paralogues puisqu'elles sont issues de la même espèce (Homo sapiens). |
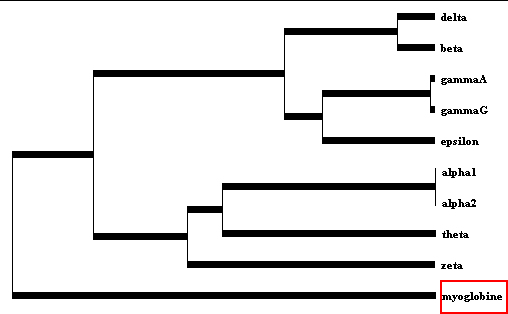
La myoglobine diffère des chaînes α et β de l'hémoglobine, davantage que celles-ci ne diffèrent l'une de l'autre. La myoglobine a divergé avant l'apparition des gènes alpha et bêta. Source figure : "La famille multigénique des globines" (ENS) |
|
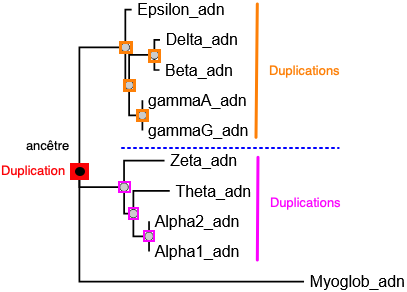
Question 2 : construction et visualisation de l'arbre aves les séquences des gènes codant les globines L'arbre phylogénétique est construit avec l'enchaînement des programmes "FastME - OneClick" (NGPhylogeny).
|
Cet arbre est semblable à l'arbre ci-dessus (ENS). On peut localiser les duplications à partir de l'ancêtre commun qui ont généré :
Source figure : NGPhylogeny |
|
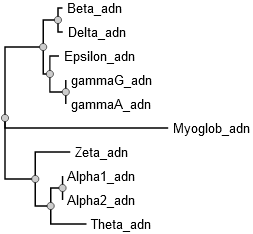
Question 2 : construction et visualisation de l'arbre aves les séquences des gènes codant les globines L'arbre phylogénétique est construit avec l'enchaînement des programmes "PhyML - OneClick" (NGPhylogeny). |
L'arbre obtenu a quasiment la même topologie que le précédent (changement de l'embranchement de l'hémoglobine ε).
Source figure : NGPhylogeny |
|
Exercice 2 : étude de l'insuline Récupération des séquences d'acides aminés d'insulines au format FASTA sur Moodle : fichier "Exercice 2 - sequences insuline". Création de l'enchaînement personnel "à la carte" des programmes de la suite logicielle NGPhylogeny pour générer l'arbre phylogénétique.
Source : NGPhylogeny |
Duplication
Spéciation
Source : Studer & Robinson-Rechavi (2009) |
|
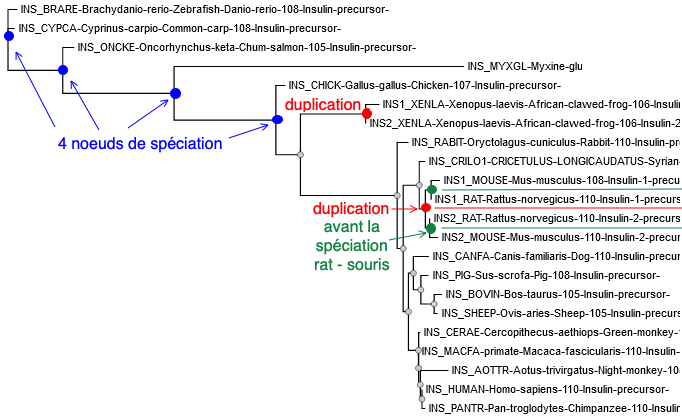
L'arbre phylogénétique de l'insuline (ci-contre) est construit avec l'option personnel "à la carte". Il y a eu 2 évènements indépendants au cours de l'évolution de la famille de l'insuline : Une duplication chez le xénope :
Une duplication chez les rongeurs :
Source figure : NGPhylogeny |
|
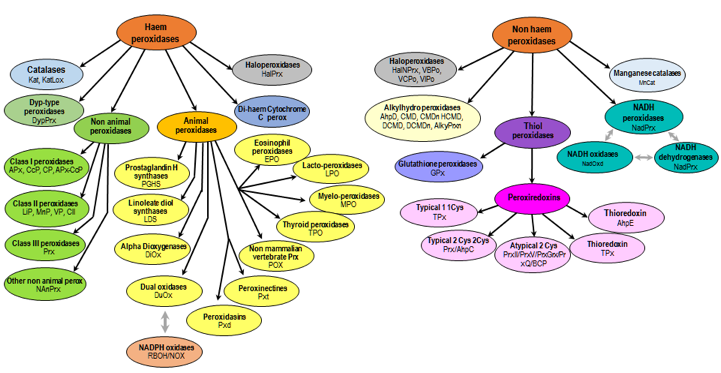
Exercice 3 : étude des peroxydases de classe III liées à la membrane Les peroxydases végétales sécrétoires ou peroxydases de classe III (EC 1.11.1.7) sont :
Exemples de fonctions : élimination du peroxyde d'hydrogène des chloroplastes et du cytosol ; oxydation de composés toxiques ; biosynthèse de la paroi cellulaire ; défense face aux blessures ; catabolisme de l'acide indole-3-acétique ; biosynthèse de l'éthylène ... Réaction enzymatique Les peroxydases sont des enzymes contenant un hème qui utilisent le peroxyde d'hydrogène comme accepteur d'électrons pour catalyser des réactions d'oxydo-réduction. La plupart des peroxydases à hème suivent le schéma réactionnel :
|
PeroxiBase (08/2018) : au moins 158 peroxydases de classe III codées par le génome du maïs (Zea mays), 155 isoenzymes dans le génome du riz (Oryza sativa), 103 isoenzymes chez Medicago truncatula et 75 isoenzymes chez Arabidopsis thaliana.
Source : PeroxiBase
Les isoenzymes :
Les isoenzymes catalysent la même réaction biochimique, généralement avec des valeurs de paramètres cinétiques différentes ou elles sont régulées différemment. |
La base de données PDB ("Protein Data Bank") Elle contient les fichiers de données de structures 3D (déterminées par différentes techniques physiques) de environ 216.000 molécules, en grande majorité des protéines.
Voir un script Python pour déterminer la position de cystéines impliquées dans des ponts disulfures à partir de fichier de la PDB. |
La base de données PROSITE C'est une base de données de domaines de protéines et de familles de protéines, regroupés sur la base des similitudes de leurs séquences : ces protéines ont généralement des fonctions identiques ou semblables et dérivent d'un ancêtre commun. Certaines régions de protéines sont mieux conservées au cours de l'évolution car elles sont importantes pour la fonction et/ou le maintien de la structure tridimensionnelle. PROSITE contient actuellement des profils ("profile") spécifiques de plus de 1000 familles ou domaines de protéines, accompagnés d'une documentation sur la structure et la fonction de ces protéines.
|
Modèle PROSITE ("PROSITE pattern") La notation PROSITE utilise le code à 1 lettre de l'IUPAC ("International Union of Pure and Applied Chemistry") avec le symbole "-" écrit entre les éléments du motif. Aller à PROSITE. Fenêtre "Search PROSITE" : entrer "heme peroxidase" => cliquer sur le lien "PDOC00394 Heme peroxidase signatures and profiles". Résultats
Lien pour accéder au logo PROSITE => cliquer sur "Retrieve the sequence logo from the alignment". |
Structure des peroxydases de classe III Leurs structures sont bien conservées.
Analyse du site actif (figure 1 de Lüthje & Martinez-Cortes (2018) "Membrane-Bound Class III Peroxidases: Unexpected Enzymes with Exciting Functions" Int. J. Mol. Sci. 19, 2876) Figures a et b : superposition du site actif de la peroxydase du raifort ("horseradish" - HRP, en bleu) et du site actif de OsPrx95 (Oryza sativa, en jaune).
Figure c : alignement de séquences multiples de peroxydases de classe III (HRP, peroxydases liées à la membrane et peroxydases solubles) généré par le programme "Clustal Omega".
|
Réponses à quelques questions
Représentation graphique de séquences alignées dite "logo" Elle traduit la conservation des résidus (nucléotides ou acides aminés) dans des régions particulières (site actifs, sites de fixation, …) de ces séquences : un logo représente donc un motif consensus.
|
Figure ci-dessous : représentation logo du site de fixation de l'hème (PROSITE PS00435) des peroxydases de classe III.
Source : PROSITE |
Figure ci-dessous : séquences du site de fixation proximal (positions moyennes 155 à 165) de l’hème de quelques peroxydases de classe III (Lüthje & Martinez-Cortes, 2018).
Expression régulière du motif consensus : |
Figure ci-dessous : représentation logo du site actif (PROSITE PS00436) des peroxydases de classe III.
Source : PROSITE |
Figure ci-dessous : séquences du site actif (positions moyennes 55 à 65) de quelques peroxydases de classe III (Lüthje & Martinez-Cortes, 2018).
Expression régulière du motif consensus : |
Visualisation de la peroxydase de Armoracia rusticana (raifort) à une résolution de 1,55 Å. Code PDB : 1W4W
|





{kind=link}