| Le repliement des protéines ("Protein folding") |
| Tweet |
|
|
1. Introduction 2. Aspect cinétique - Paradoxe de Cyrus Levinthal 3. Aspect thermodynamique du repliement 4. L'entonnoir des paysages énergétiques 5. Rôle du solvant : l'eau 6. Quelques modèles du repliement |
7. Les moyens pour approcher le problème du repliement 8. Expériences de calcul partagé pour la simulation du repliement 9. Méthodes de simulations de la dynamique moléculaire du repliement 10. Apport de l'apprentissage profond à la prédiction de la structure 3D 11. Inactivation pharmacologique de protéine par ciblage d'intermédiaire du repliement 12. Liens Internet et références bibliographiques |
| 1. Introduction
Le processus par lequel la chaîne polypeptidique d'une protéine acquière une structure tridimensionnelle s'appelle le repliement : ce processus n'est pas encore décrypté.
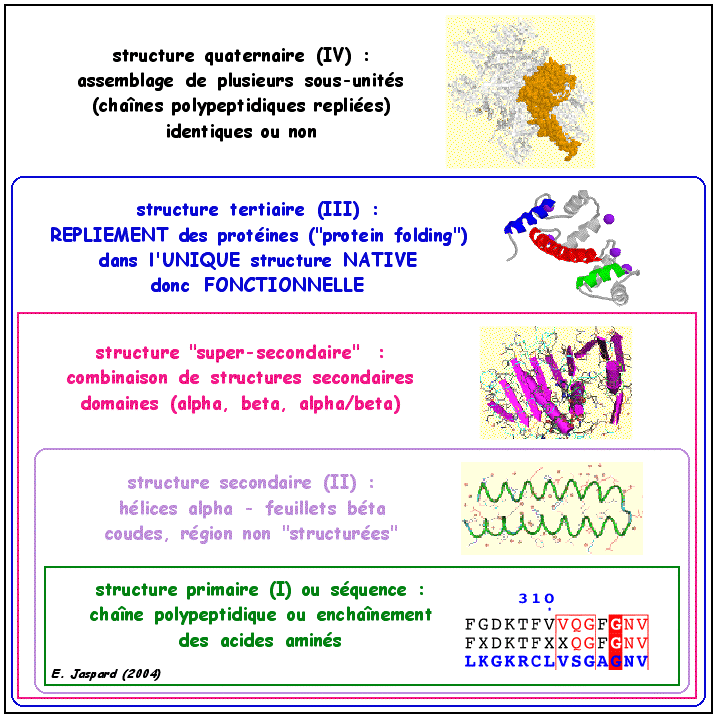
La situation est beaucoup plus complexe que dans le cas des acides nucléiques. En effet, chacune des milliers de protéines qui existent dans une cellule doit reconnaitre spécifiquement un (ou quelques) ligand(s). Cette reconnaissance s'effectue grâce à de subtiles interactions entre la structure tridimensionnelle de la protéine et celle(s) du (des) ligand(s). Une telle diversité d'interactions aussi précises ne peut être obtenue qu'avec des structures de protéines extrêmement variées et irrégulières. La figure ci-dessous montre la "hiérarchie" des 4 niveaux de structures des protéines.
Christian Anfinsen (Prix Nobel 1972) a montré que, dans un environnement approprié : toute l'information nécessaire au repliement d'une protéine dans sa structure native (donc fonctionnelle) est contenue dans sa séquence primaire (l'enchaînement des acides aminés). Pour rendre compte de la complexité du processus du repliement, on peut mentionner :
Figure ci-dessous : représentation schématique des liaisons au sein d'une protéine repliée. A ces liaisons s'ajoutent les interaction avec le milieu ambiant.
|
| Valeurs d'énergie des liaisons qui s'établissent au sein d'une protéine et qui stabilisent sa structure native. | |
| liaison | énergie (kcal/mole) |
| covalentes : pont disulfure | 90 |
| électrostatiques | 3 |
| hydrogène | 3 - 7 / la valeur dépend du donneur - accepteur |
| contacts de van der Waals | 0,1 - 1 |
| interactions hydrophobes | pas une liaison au sens strict, mais rôle clé dans la stabilisation |
| Facteur additionnel : les propriétés physico - chimiques et les contraintes stériques de la liaison peptidique (la chaîne des carbones α) - les angles Φ (phi), Ψ (psi) et ω (omega). | |
Pour mesurer la compléxité de ce (ou ces) mécanisme(s), rappelons que :
|
|
2. Aspect cinétique - Paradoxe de Cyrus Levinthal Le nombre de conformations d'une chaîne polypeptidique de n acides aminés (susceptibles d'adopter s structures secondaires) est sn. Autre ordre de grandeur : 100 acides aminés => 99 liaisons peptidiques => 198 angles φ et ψ => 3 conformations stables pour chacun de ces angles => 3198 conformations possibles. Hypothèse que le repliement d'une protéine suit un processus au hasard : une protéine contenant n = 100 acides aminés / adoptant s = 13 structures secondaires / temps d'exécution de chaque conformation = 1 psec => temps pour explorer toutes les conformations = 1085 secondes, c'est-à-dire 3 1017 ... siècles ! En conséquence, un processus du repliement opèrant uniquement selon une recherche au hasard de la structure de plus basse énergie (la plus stable) est donc absolument impossible du point de vue cinétique et totalement incompatible avec les échelles de temps de la vie biologique et avec la vitesse des évènements du repliement qui s'échelonnent de la micro-seconde à la milli-seconde. C'est le paradoxe de Cyrus Levinthal ("How to fold graciously", 1969). Les études de la cinétique du repliement ont montré l'existence de structures intermédiaires instables. La grande difficulté a toujours résidé dans l'isolement (et donc la caractérisation) de ces intermédiaires peu peuplés et trés labiles (temps de demi-vie trés courts). Cependant, les données accumulées montrent actuellement que le le repliement correspond à une ou des étape(s) initiale(s) rapide(s) suivie(s) de réarrangements structuraux plus lents (ajustements conformationnel locaux) Une théorie a été développée par Alan Fersht : elle porte sur l'aspect cinétique du repliement (via les équilibres entre intermédiaires et/ou états globaux : état natif, états dénaturés, états structuraux intermédiaires). |
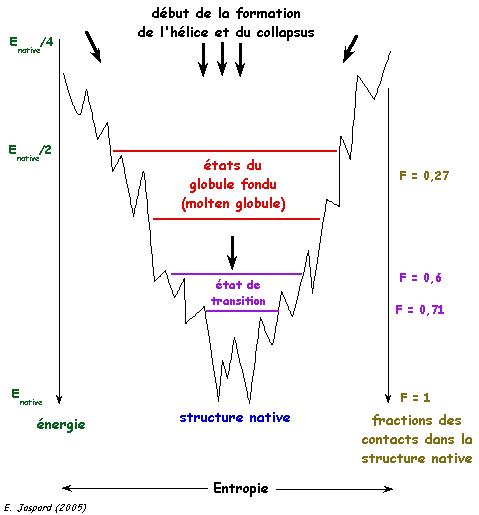
3. Aspect thermodynamique du repliement Les états dénaturés (l'ensemble des conformations qu'une chaîne polypeptidique adopte quand elle n'est pas repliée ou est incorrectement repliée) et l'état natif (unique) d'une protéine ont des niveaux d'énergie trés proches. En revanche, leur différence d'entropie (le nombre des configurations possibles dans chaque état) est trés importante. Du point de vue statistique, on peut donc considérer que l'état natif est largement improbable (c'est une autre illustration du paradoxe de Levinthal). Les données actuelles sont en faveur d'un processus du repliement contrôlé thermodynamiquement. La formation de la structure native (le fond de l'entonnoir dans la figure ci-dessous) s'accompagne d'une diminution de l'entropie. [Wolynes et al. (1995) Science 267, 1619 - 1620].
Figure adaptée de Benhabilès et al. (2000) Les intermédiaires structuraux dont la formation est sous contrôle cinétique aboutissent à la formation de la structure native. Récemment la question a été posée de savoir si l'hypersurface du potentiel qui dirige le repliement est constituée de minima multiples ou s'il s'agit d'un système percolatoire dans lequel ces minima seraient des points de selle. |
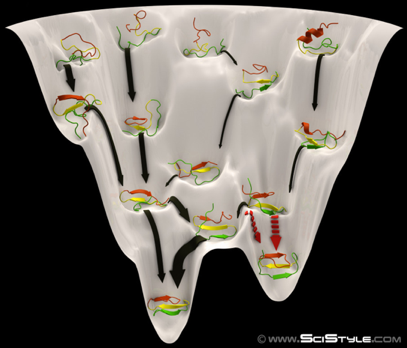
4. L'entonnoir des paysages énergétiques ("Folding energy landscape") Le nombre de conformations adoptables par une chaîne polypeptidique en train de se replier est astronomique. La forme de l'espace de ces conformations (ou paysage énergétique) est représentée par une fonction mathématique qui décrit les énergies libres intramoléculaire et de solvatation d'une protéine en fonction des degrés de liberté microscopiques. L'une des grandes avancées théoriques dans la résolution du problème du repliement d'une chaîne polypeptidique, a été de quantifier la fonction de partition de mécanique statistique, dont une composante clé est la densité d'état ("Density Of States" - DOS) : le nombre de conformations à chaque niveau énergétique. Dans les cas simples, le logarithme de DOS est l'entropie conformationnelle. De telles valeurs d'entropie n'ont pu être estimées via des modèles tenant compte de tous les atomes constitutifs d'une protéine car cela aurait requis des temps et des méthodes de calculs astronomiques. Des modèles simplifiés de la chaîne polypeptidique en cours de repliement ont donc été développés. L'une des conclusions les plus importantes est que les protéines sont caractérisées par un paysage énergétique en forme d'entonnoir ("funnel-shaped energy landscape"). Les chaînes polypeptidiques adoptent beaucoup d'états de grande énergie et peu de faible énergie. Une chaîne polypeptidique résout le grand problème d'optimisation globale comme une série de petits problèmes d'optimisation locale : elle assemble des fragments peptidiques préalablement structurés et aboutit ainsi de proche en proche à sa structure native.
Source : Behance |
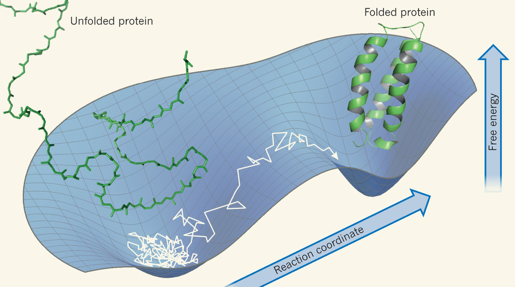
Il y a une distinction capitale à faire entre le repliement d'une protéine et une réaction chimique classique simple.
En conséquence, un diagramme à 1 ou 2 dimension(s) du contour réactionnel du repliement ne peut décrire cette réduction drastique du nombre de conformations. En revanche, une forme en entonnoir :
Source : Schuler & Clarke (2013) Les structures des protéines oscillent entre différentes conformations pour exécuter leurs tâches sophistiquées. Le mouvement aléatoire des molécules d'eau autour des protéines fournit un réservoir inépuisable de "coups" thermiques qui agissent comme des moteurs moléculaires de la dynamique conformationnelle des protéines. Il semble cependant que, dans certains cas, les phénomènes de friction au sein d'une protéine soit l'entrave dominante à sa dynamique moléculaire. |
|
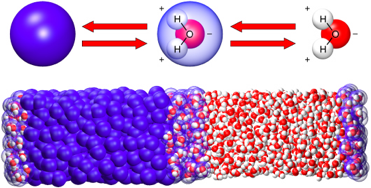
Le solvant naturel (dans la cellule) des protéines est l'eau :
Le repliement est un processus dynamique :
Remarque : il faut distinguer
Simulation de la molécule d'eau : lien entre le modèle appelé à grain grossier ("coarse grained" - à gauche) et le modèle appelé tout-atome ("all-atom" - à droite).
Source : Praprotnik et al. (2007) |
6. Quelques modèles du repliement Les molécules biologiques ont un large éventail d'échelles de temps au cours desquelles des processus spécifiques ont lieu :
|
| Modèle | Description | Références |
| nucléation - condensation | Il y a une étape de nucléation
suivie par une propagation rapide de la structure. C'est un modèle qui prend en compte le caractère coopératif du repliement. |
Zimm & Bragg (1959) J. Chem. Phys.
31, 526 - 535 Wetlaufer (1973) Proc. Natl. Acad. Sci. USA 70, 697 - 701 |
| diffusion - collision | La nucléation intervient en plusieurs
points de la chaîne polypetidique. Ces noyaux diffusent et coalescent. On aboutit à des microstructures natives. Le repliement est une succession d'étapes de diffusion - collision. |
Karplus & Weaver (1994) Protein Sci. 3, 650 - 668 |
| repliement séquentiel et hiérarchique | Plusieurs segments de structures sont formés
et assemblés à différents niveaux suivant un
chemin du repliement unique (Baldwin, 1975). Schulz (1977) a proposé que ce repliement soit hiérachique : la nucléation est suivie par la formation de structures super - secondaires puis par celle des domaines (voire du monomère). |
Baldwin (1975) Annu. Rev. Biochem.
44, 453 - 475 Schulz (1977) Angew. Chem. 16, 23 - 32 |
| modèle modulable du repliement | Les domaines d'une protéine sont les
unités du repliement (Wetlaufer, 1981). Ils se replient indépendamment et des intermédiaires du repliement (les modules structuraux) sont formés et s'assemblent pour aboutir à la structure native (Chotia, 1984). |
Wetlaufer (1981) Adv. Protein Chem. 34, 61 - 92 Chotia (1984) Annu. Rev. Biochem. 53, 537 - 572 |
| modèle d'effondrement (collapse) | Le premier évènement du repliement
est un effondrement hydrophobe avant la formation des structures secondaires. Cet effondrement conduit à la stabilisation de la strucure native. |
Levitt & Warshel (1975) Nature 253, 693 - 698 |
| fermeture éclair hydrophobe | La formation de segments de structures secondaires est simultanée avec l'effondrement hydrophobe. | Dill et al. (1993) Proc. Natl. Acad. Sci. USA 90, 1942 - 1946 |
| homopolymère et extension de ce modèle |
Modèle qui décrit
certains aspects universels des protéines comme les régions
spatialement structurées en hélice ou en feuillet. La
protéine est alors considérée comme un polymère
où tous les maillons sont identiques (homopolymère). Modèle qui va au-delà du modèle simple d'homopolymère : tous les acides aminés sont identiques à l'exception de la glycine et de la proline. |
aller au site |
| autres modèles |
Les modèles de verres de spin
peuvent être appliqués au repliement en décrivant les protéines comme
des hétéropolymères aléatoires. Une protéine serait un objet quantique dont on pourrait expérimentalement observer les états excités selon différents modes. |
----- |
| les foldons | Certains résultats expérimentaux (échanges isotopiques à l'équilibre ou cinétique) sont en faveur de l'hypothèse que les protéines sont des objets à états multiples plutôt qu'à 2 états. Elles seraient composées de blocs séparés et coopératifs appelés foldons, unités capables de se replier et de se déplier à plusieurs reprises même en conditions natives.
|
Efimov et al. (1994) |
Simulations de la dynamique moléculaire ("molecular dynamics simulations") du repliement :
|
voir une simulation | |
7. Les moyens pour approcher le problème du repliement On dispose de moyens techniques et bioinformatiques mais aussi d'un grand nombre de bases de données ("Protein Data Bank", SCOP, CATH, PFAM, CDD, ...) pour approcher une solution la plus complète du problème du repliement de n'importe quelle protéine. Parmi les techniques, on peut citer entre autres :
Bien que la connaissance actuelle de l'ensemble des forces et des mécanismes microscopiques qui gouvernent le repliement soit largement incomplète, cela n'a pas empêché l'émergence de nouvelles méthodes de conception de protéines :
De plus, de nouveaux polymères appelés "foldamères" permettent d'obtenir des structures hélicoïdales compactes avec des constituants non biologiques :
Ces foldamères ont trouvé des applications en biomédecine où ils sont utilisés :
|
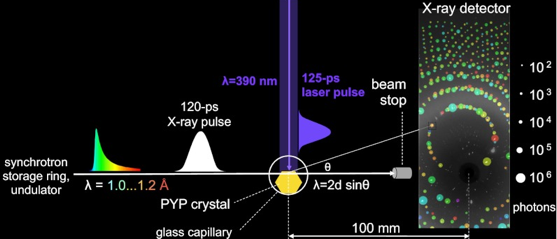
Etude des intermédiaires du repliement La caractérisation d'intermédiaires structuraux du repliement reste l'une des difficultés majeures de l'étude du repliement. En effet, le temps de demi-vie de ces intermédiaires est souvent très court (de l'ordre de la ps ou ns) et leur nombre est grand. De nouvelles technologies permettent d'aborder ce domaine avec des résultats prometteurs. L'une d'entre elle est la ligne de faisceau BioCARS 14-BID ("BioCARS 14-IDB beamline at the Advanced Photon Source"). Cette technologie a été employée pour suivre les changements structuraux dans des cristaux de protéine avec une résolution spatiale atomique et une résolution en temps de 150 ps : ils ont permis de suivre le photocycle réversible de la protéine jaune photoactive ("Photoactive Yellow Protein"- PYP) après la photoisomérisation trans -> cis de son chromophore acide p-coumarique (pic d'absorbance dans un cristal P63 : 447 nm). Schématiquement, des instantanés (de l'ordre de la ps) de la structure de PYP ont été acquis à l'aide de la méthode "pompe-sonde":
Source : Schotte et al. (2012)
L'impulsion de rayons X (12 keV) utilisée étant poly-chromatique, des milliers de réflexions sont capturées en une seule image (spot de 80 × 40 μm2) sans avoir à tourner le cristal. Cette approche augmente considérablement la vitesse à laquelle les données de diffraction sont acquises, d'où une augmentation de la résolution dans le temps. Les informations nécessaires pour déterminer la structure de la protéine sont contenues dans les intensités relatives des taches de diffraction observées. Cependant, les informations structurales contenues dans une seule image de diffraction étant incomplètes, cette méthode nécessite des mesures répétées avec des orientations multiples du cristal pour produire un ensemble complet de données. Dans cette étude, des données de diffraction de 9 très gros (pour atténuer les effets néfastes des radiolésions lors de la collecte de données) cristaux différents dans 41 orientations ont été combinées pour produire des cartes de densité électronique résolues dans le temps. |
|
8. Expériences de calcul partagé pour la simulation du repliement Les échelles de temps du repliement s'étendent de l'ordre de 10-11 s à 10-3 s. Certaines sont trop courtes pour être analysées avec les technologies actuelles. Elles peuvent être simulées par ordinateur. Depuis quelques années, des expériences de calcul partagé (grille ou "grid" : ensembles d'ordinateurs en réseau) ont lieu dans le but est de prévoir la structure d'une protéine.
CASP ("Critical Assessment of Techniques for Protein Structure Prediction") En 1994, John Moult a inventé CASP ("Critical Assessment of Techniques for Protein Structure Prediction"), un test "à l'aveugle" ouvert à l'ensemble de la communauté scientifique pour prédire la structure inconnue de protéines.
Voir ci-dessous : "Apport de l'apprentissage profond". |
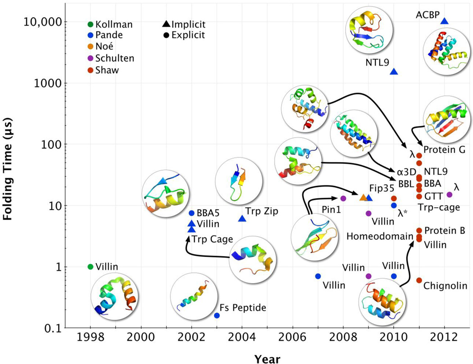
9. Méthodes de simulations de la dynamique moléculaire du repliement Les simulations de la dynamique moléculaire ("Molecular Dynamics Simulations" - MDS) du repliement des protéines (quantitativement précises) constituent l'une des approches les plus récentes et les plus prometteuses. Au cours de la dernière décennie, les tailles des systèmes biologiques étudiés et les échelles de temps du repliement accessible aux méthodes de simulations ont augmenté de manière exponentielle (figure ci-dessous).
Source : Lane et al. (2013) Ce gain a été obtenu grâce à des progrès sur trois fronts principaux : une parallèlisation des codes de MDS, du matériel informatique spécialisé et de plus en plus performant (exemple : Anton - superordinateur pour calculs massivement parallèles construit par Shaw Research) et l'analyse statistique de trajectoires multiples indépendantes. Evolution des capacités de simulations de systèmes aussi complexes qu'une protéine et son environnement aqueux :
Voir : San Diego Supercomputer Center et Computational Biochemistry Group (Kästner Group - Stuttgart). |
10. Apport de l'apprentissage profond à la prédiction de la structure 3D L'analyse de co-évolution ("residue-residue coevolutionary") est basée sur l'observation selon laquelle 2 résidus d'acides aminés en contact (ou proches dans l'espace selon un seuil de distance, par exemple 8 Å) doivent co-évoluer pour maintenir ce contact. En d'autres termes, si un acide aminé est muté en résidu chargé positivement, l'acide aminé avec lequel il est en contact doit être muté en résidu chargé négativement. Depuis le concours CASP11 et CASP12, les scores de co-évolution sont de plus en plus utilisés comme entrées des méthodes d'apprentissage profond afin d'améliorer la prédiction des contacts et des distances ("inter-residue contacts and distances"). |
| Exemples d'outils de prédiction de la structure 3D des protéines | |
| Outils | Principe |
| CASP13 | Les prédicteurs ont calculé la topologie de repliement de la chaîne polypeptidique avec une résolution < 3 Å par rapport à la structure déterminée expérimentalement. |
| AlphaFold | Conceptuellement, AlphaFold était à l'origine un réseau de neurones pour prédire les distances probables entre les paires d'acides aminés et les angles de chaque liaison peptidique reliant les résidus d'acides aminés. Ces 2 prédictions ont ensuite été intégrées à un score avec le "score2" du logiciel de modélisation Rosetta. AlphaFold utilise la distribution entière des distances 2 à 2 entre résidus d'acides aminés comme une fonction potentielle statistique (spécifique de la protéine) qui est directement minimisée par la descente de gradient pour obtenir le repliement de la protéine. AlphaFold est au coeur de la base de données (système de prédiction) AlphaFold DB. |
| RaptorX - Contact | Serveur Web qui applique un réseau de neurones convolutifs ultra-profond pour la prédiction des cartes de contacts et la prédiction de matrices de distances de séquences protéiques sans l'utilisation de modèles. RaptorX utilise uniquement la moyenne et la variance de la distribution prédite. |
| iFeature | Package Python et un serveur Web pour l'extraction et la sélection de caractéristiques à partir de séquences de protéines et de peptides. |
| iLearn | Plate-forme intégrée et "méta-apprenant" pour l'ingénierie des caractéristiques, l'analyse par apprentissage automatique et la modélisation des données de séquences d'ADN, d'ARN et de protéines. |
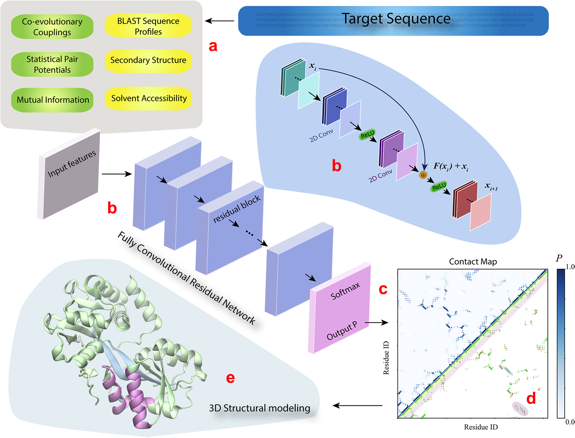
| DESTINI | (a) Les caractéristiques 1D (bulles jaunes en haut à gauche, figure ci-dessous) et 2D (bulles verts) de la séquence de la protéine analysée ("Target sequence") sont extraites à partir des données génomiques et structurales. (b) Ces caractéristiques constituent les entrées ("Input features") d'un réseau convolutif composé de plusieurs blocs résiduels identiques (le détail de l'architecture est décrit dans la bulle bleue claire). (c) La couche de sortie du réseau est une couche d'activation Softmax qui génère le score de probabilité ("Output P") pour chaque paire de résidus de la séquence cible. (d) La sortie est affichée sous forme d'une carte de contacts prédits ("Contact map") avec les scores de probabilité (partie gauche - haute de la carte) et une comparaison des contacts prédits (P > 0,5) avec les contacts véritables (partie droite - basse). (e) Les contacts prédits sont ensuite utilisés pour générer le modèle 3D de la protéine analysée. Les couleur des contacts entre les hélices α et les brins β sont identiques à celles de la carte des contacts (d).
Source : Gao et al. (2019) |
| MULTICOM | Il s'appuie sur 3 concepts principaux : (i) la prédiction de la distance de contacts avec des réseaux de neurones convolutifs; (ii) la modélisation sans modèle (ab initio) dirigée par la distance; (iii) le classement du modèle de la protéine renforcé par l'apprentissage profond et la prédiction des contacts. |
| SPIDER2 | Prédiction des erreurs dans les valeurs prédites des angles de torsion de la chaîne polypeptidique, de l'accessibilité au solvant, des contact . |
| Early Folding Residue Predictor | Le repliement des protéines est un processus qui s'effectue par étapes : certains résidus d'acides aminés semblent initier ce processus ("early folding residues"). Certaines données expérimentales concernant ces résidus sont fournies par la base de données Start2Fold mais pour la plupart des structures protéiques, aucune donnée n'est disponible. Early Folding Residue Predictor prédit ("Generalized Matrix Learning Vector Quantization") la position de ces résidus à partir de structures protéiques (PDB). |
Autres outils : UniCon3D (CASP11) - DNCON2 (CASP 12) - MetaPSICOV2 (CASP 12, utilise les données de PDB70 et Uniref100) - DeepCov, ... |
|
11. Inactivation pharmacologique de protéine par ciblage d'intermédiaire du repliement Les progrès dans le domaine de la simulation des processus biochimiques permettent d'étudier les mécanismes impliqués dans la régulation des protéines avec des modèles à un niveau de résolution atomistique. Ces techniques informatiques sont à l'origine d'une méthode appelée inactivation pharmacologique de protéine par ciblage d'intermédiaire du repliement ("Pharmacological Protein Inactivation by Folding Intermediate Targeting" - PPI-FIT).
Génération de chemins de repliement avec la méthode du biais fonctionnel ("bias functional method")
Ces résultats renforcent l'idée que la teneur des protéines peut être modulée en agissant sur leurs trajectoires de repliement, soulignant ainsi un rôle possible des intermédiaires du repliement dans la régulation de la synthèse des protéines. |
| 12. Liens Internet et références bibliographiques |
| "Introduction à la structure des protéines" - C. Branden & J. Tooze (1996) - ed. De Boeck Université | |
Institute for Protein Design - University of Washington Critical Assessment of Techniques for Protein Structure Prediction |
|
| AlphaFold DB : système de prédiction de la structure des protéines du protéome humain et de 20 autres organismes clés, basé sur l'intelligence artificielle. C'est une initiative de DeepMind et de EMBL European Bioinformatics Institute (EMBL-EBI). |
AlphaFold DB |
|
Expériences de calcul partagé dont le but est de prévoir la structure d'une protéine
|
|
|
"FoldIt" : "jeu vidéo" développé par les membres du laboratoire de D. Baker - Université de Washington "RosettaAThome": détermination de structures tridimensionnelles avec temps de calcul partagé Chow et al. (2005) "REFOLD: An analytical database of protein refolding methods" Protein. Expr. Purif. 46, 166 - 171 Khatib et al. (2011) "Algorithm discovery by protein folding game players" P.N.A.S. 108, 18949 - 18953 |
|
|
Anfinsen et al. (1961) Proc. Natl. Acad. Sci. USA 47, 1309 - 1314 Levinthal C. (1968) "Are there pathways for protein folding ?" J. Chem. Phys. 65, 44 - 45 Levinthal, C. (1969) "How to fold graciously" In "Mossbauer spectroscopy in biological systems" (eds. P. DeBrunner et al.), 22 - 24. University of Illinois Press, Allerton House, Monticello, IL. Efimov et al. (1994) "Fibritin Encoded by Bacteriophage T4 Gene wac has a Parallel Triple-stranded α-Helical Coiled-coil Structure" J. Mol. Biol. 242, 470 - 486 |
|
Benhabilès et al. (2000) "Les mécanismes de repliement des protéines solubles" Biotechnol. Agron. Soc. Environ. 4, 71 - 81 Grantcharova et al. (2001) "Mechanisms of protein folding" Curr. Opin. Struct. Biol. 11, 70 - 82 Fersht, A. R. (2002) "On the simulation of protein folding by short time scale molecular dynamics and distributed computing" PNAS 99, 14122 - 14125 |
|
|
Praprotnik et al. (2007) "Adaptive resolution simulation of liquid water" J. Phys.: Condens. Matter 19 No 29 Dill et al. (2008 ) "The Protein Folding Problem" Annu. Rev. Biophys. 37, 289 - 316 Ferreon et al. (2011) "Protein folding at single-molecule resolution" Biochim. Biophys. Acta 1814, 1021 - 1029 |
|
|
Vallée-Bélisle & Michnick (2012) "Visualizing transient protein-folding intermediates by tryptophan-scanning mutagenesis" Nature Struct. Mol. Biol. 19, 731 - 736 Schotte et al. (2012) "Watching a signaling protein function in real time via 100-ps time-resolved Laue crystallography" Proc. Natl. Acad. Sci. USA 109, 19256 - 19261 Yamada et al. (2013) "Snapshots of a protein folding intermediate" PNAS 110, 1606 - 1610 Schuler & Hofmann (2013) "Single-molecule spectroscopy of protein folding dynamics-expanding scope and timescales" Curr. Opin. Struct. Biol. doi: 10.1016 |
|
Jung et al. (2013) "Volume-conserving trans–cis isomerization pathways in photoactive yellow protein visualized by picosecond X-ray crystallography" Nature Chem. 5, 212 - 220 Schuler & Clarke (2013) "Biophysics: Rough passage across a barrier" Nature 502, 632 - 633 Lane et al. (2013) "To Milliseconds and Beyond: Challenges in the Simulation of Protein Folding" Curr. Opin. Struct. Biol. 23, 58 - 65 |
|
|
AlQuraishi M. (2019) "AlphaFold at CASP13" Bioinformatics doi: 10.1093/bioinformatics/btz422 Gao et al. (2019) "DESTINI: A deep-learning approach to contact-driven protein structure prediction" Sci. Rep. 9, 3514 Bittrich et al. (2019) "Application of an interpretable classification model on Early Folding Residues during protein folding" BioData Min. 12, 1 |
|
|
Spagnolli et al. (2021) "Pharmacological inactivation of the prion protein by targeting a folding intermediate" Commun. Biol. 4, 62 Tunyasuvunakool et al. (2021) "Highly accurate protein structure prediction for the human proteome" Nature 596, 590 - 596 |
|
![]()