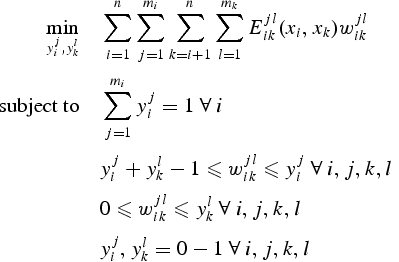| Détermination et prédiction des structures des protéines |
| Tweet |
|
|
1. Introduction 2. Classifications hiérarchiques des structures des protéines 3. Les techniques pour déterminer - analyser les structures des protéines
|
4. La mécanique et la modélisation moléculaires 5. Les méthodes "ab initio"
6. Liens Internet et références bibliographiques |
1. Introduction Les protéines se replient dans l'espace pour adopter une structure tridimensionnelle native unique qui leur confère leur propriétés biologiques. Il y a un nombre important de repliements des protéines observés dans la nature.
L'évolution du nombre de nouveaux repliements découverts lors des dernières années indique :
On ne sait pas si les repliements non encore observées sont physiquement impossibles ou si elles n'ont pas encore été "testées" par le processus évolutif ou caractérisées par les biologistes structuraux. Remarque : certaines protéines sont dites intrinsèquement non structurées et certaines nécessitent une aide au repliement par des protéines chaperonnes. Quoi qu'il en soit, ces protéines adoptent à un moment ou un autre une structure tridimensionnelle unique. |
a. Les 3 grands types de protéines α. Les protéines fibreuses : ce sont des protéines allongées dont les éléments de structure secondaire sont les structures dominantes. Exemple : la kératine. β. Les protéines membranaires :
γ. Les protéines globulaires :
|
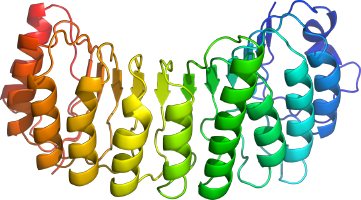
b. Terminologies liées aux structures des protéines Domaines : ce sont des unités fonctionnelles et/ou structurales distinctes des protéines. Ils sont en général responsables d'une fonction ou d'une interaction particulière et contribuent à la fonction de la protéine. Les domaines peuvent en général se replier de manière indépendante. Figure ci-dessous : les motifs répétés "Leucine-rich repeat" (LRR) du domaine N-terminal de RanGAP1 ("Ran GTPase-activating protein 1"). Type de repliement : superhélice [β-α] de pas droit ("right-handed beta-alpha superhelix").
Source : SCOP Motif ("motif") :
|
| Courts motifs (2 à 6 acides aminés) stabilisés par des liaisons hydrogène, trouvés dans les protéines | ||
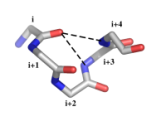
| alpha-beta-motif | asx-motif | Exemple de définition : motif α-β (figure ci-dessous)
Motif fréquent dans les hélices α en position C- et N-terminales. Les motifs α-β de pas gauche sont rares. Caractéristiques structurales :
|
| asx-turn-iL | asx-turn-iR | |
| asx-turn-iiL | asx-turn-iiR | |
| beta-bulge | ----- | |
| beta-bulge-loop-5 | beta-bulge-loop-6 | |
| beta-turn-iL | beta-turn-iR | |
| beta-turn-iiL | beta-turn-iiR | |
| gamma-turn-classic | gamma-turn-inverse | |
| nest-LR | nest-RL | |
| niche-3R | niche-3L | |
| niche-4L | niche-4L | |
| schellmann-loop-6 | schellmann-loop-7 | |
| st-staple | st-motif | |
| st-turn-iL | st-turn-iR | |
st-turn-iiL |
st-turn-iiR | |
| PDBeMotif: interface Web pour la recherche de motifs selon divers critères dans les protéines de la PDB. | ||
Patron ("pattern") : courte séquence en acides aminés essentiels à la fonction d'une protéine (site de fixation, site actif, ...). Ils sont mis en évidence par alignements multiples de séquences de protéines ayant des fonctions comparables. Si les séquences des "pattern" ne sont pas exactes, on les exprime sous forme d'expression régulière (exemple : [FY]-x-E-x(4)-{ILV}). Empreinte ("fingerprints") : ensemble de courts motifs conservés (mis en évidence par alignements multiples de séquences). |
|
c. La "Protein DataBank" La base de données mondiale recueil des structures tri-dimensionnelles des macromolécules biologiques est la Protein Data Bank (PDB). Elle contient environ 182.000 structures de protéines, d'acides nucléiques (ADN et ARN) et de complexes ribonucléoprotéiques, déterminées par différentes techniques :
Les 10 organismes les plus représentés sont Homo sapiens, Escherichia coli, Mus musculus, Saccharomyces cerevisiae, Bos taurus, Gallus gallus, Sus scrofa, Thermus thermophilus, ... |
2. Classifications hiérarchiques des structures des protéines La base de données SCOP ("Structural Classification of Proteins") - MRC Laboratory of Molecular Biology - Cambridge - Angleterre Sa classification est basée sur la similarité des structures et des séquences en acides aminés des domaines structuraux des protéines. L'unité de classification est le domaine d'une protéine. Pour les auteurs de cette classification, la définition de domaine SCOP se base sur le fait que les petites protéines ne contiennent généralement qu'un domaine. Exemple : l'hémoglobine (structure α2β2) est considérée comme ayant 2 domaines SCOP, un domaine α et un domaine β. Attention : ici la terminologie "α" et "β" n'a rien à voir avec une hélice α ou un feuillet β. Les niveaux de la classification SCOP :
Exemple de classification SCOP (les liens renvoient vers les données au niveau hiérarchique considéré de la classification) :
|
| Comparaison des terminologies SCOP et CATH ("Class Architecture Topology Homology") | |
| SCOPe : > 1200 repliements (septembre 2021) | Base de données CATH |
Classe |
Classe : essentiellement alpha, essentiellement beta, (alpha/beta) mélangé |
Exemple de classes de protéines et nombre de repliements
|
Architecture : classification selon la conformation globale, en ignorant toute connectivité |
| Superfamille | Topologie ("Topology") : groupes de repliements (forme et connectivité) |
| Famille | Superfamille homologue ("Homologous Superfamily") |
| Domaines : définition manuelle | Domaines : définition automatique |
Bases de données liées à SCOP
Les autres bases de données de classification SMART : "Small motif database" / PRODOM : "Protein domain database" / InterPro : "Databases of protein families and domains" / HOMSTRAD : "Homologous structure alignment database" Exemples de programmes et de serveurs d'alignement de structures SSAP ("Sequential Structure Alignment Program") / DALI / STRUCTAL ("Structural Alignment Server") / VAST ("Vector Alignment Search Tool" - NCBI) / LSQMAN / SSM ("PDBeFold"). |
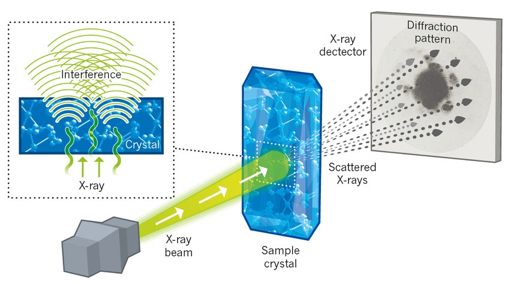
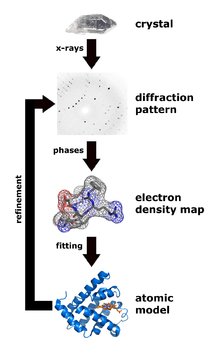
3. Les techniques pour déterminer - analyser les structures des protéines Il n'y a pas une technique meilleure qu'une autre. Elles ont toutes leur spécificité avec leurs avantages et leurs inconvénients. Les meilleurs modèles structuraux de macromolécules biologiques ou de complexes biologiques résultent de combinaisons de données obtenues par plusieurs de ces techniques. a. La diffraction des rayons X Voir le principe de la diffraction des rayons X. L'histoire raconte qu'en skiant dans les Alpes en 1912, le physicien allemand Max von Laue énonça à ses collègues une idée novatrice : il postulait que les rayons X en passant au travers d'un cristal seraient réfléchis par les centres atomiques du réseau cristallin et interfèreraient entre eux pour créer un diagramme de diffraction.
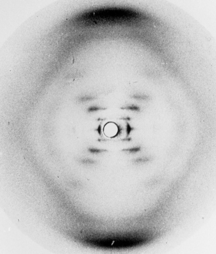
Source : N. Jones (2014) L'idée de von Laue était correcte et en 1914, il a reçu le prix Nobel de physique « pour sa découverte de la diffraction des rayons X par les cristaux ». En 2012, on a fêté le 100ème anniversaire de la loi énoncée par Lawrence Bragg : n . λ = 2 d sin θ. Figure ci-dessous : image radiographique de l'ADN obtenue en 1952 par Rosalind Franklin (appelée photo 51). Ces photographies ont été déterminantes pour l'élucidation de la structure en double hélice de l'ADN par J. Watson, F. Crick, et M. Wilkins en 1953 (Prix Nobel en 1962).
Source : King's College London R. Lefkowitz & B. Kobilka ont reçu le prix Nobel de Chimie en 2012 pour leurs travaux sur la détermination de la structure des RCPG. La cristallographie étudie les macromolécules sous forme cristalline à l'échelle atomique : c'est actuellement la technique la plus résolutive qui peut être inférieure à 1 Å.
Source : Wikipedia L'état cristallin est défini par un caractère périodique et ordonné à l'échelle atomique ou moléculaire. Ce caractère périodique est appelé la maille élémentaire. La cristallogénèse est la formation d'un cristal, soit en milieu naturel, soit de façon expérimentale. C'est le passage d'un état désordonné liquide à un état ordonné solide, contrôlé par la température, la pression, le temps d'évaporation et des lois cinétiques complexes :
Des automates permettent maintenant de tester en parallèle des centaines de conditions physico-chimiques de cristallogénèse. La plupart des substances minérales et des petites molécules organiques cristallisent facilement et les cristaux obtenus sont en général sans défaut. En revanche les macromolécules biologiques, comme les protéines (a fortiori les protéines membranaires), sont souvent très difficiles à cristalliser. Techniques de pointe pour générer des rayons X :
La diffraction des rayons X par des monocristaux ("X-ray single-crystal diffraction" - SCD) a une limitation évidente : les molécules cibles doivent être obtenues comme des monocristaux. En 2013, un protocole d'analyse SCD ne nécessitant pas la cristallisation de l'échantillon a été développé (Inokuma et al., 2013):
|
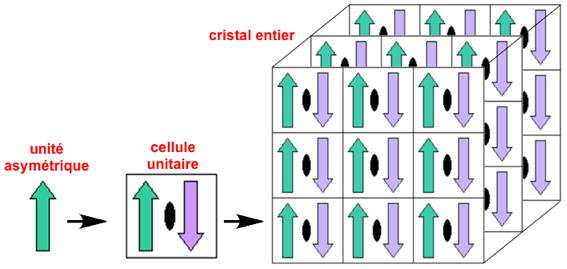
Cristallographie : unité asymétrique d'une maille cristalline et cellule unitaire L'unité asymétrique est le plus petit volume d'une structure cristalline (maille cristalline) auquel les opérations de symétrie du groupe d'espace du cristal considéré peuvent être appliquées afin de reconstituer la cellule unitaire ("unit cell" - l'unité de répétition du cristal) complète. Quand la cellule unitaire est répliquée dans les 3 dimensions, on reconstitue le cristal entier. Exemple : l'unité asymétrique (flèche verte vers le haut) est tournée de 180 degrés autour d'un axe de symétrie cristallographique double (ovale noir). On obtient une copie (flèche violette vers le bas) : ces deux flèches constituent la cellule unitaire. Celle-ci est alors répliquée par translation dans les 3 directions pour former le cristal tridimensionnel.
Source : PDB Remarque : le volume de l'unité asymétrique est donc inférieur à celui de la maille du cristal sauf pour le groupe d'espace triclinique P1 dont l'unité asymétrique a un volume égal à celui de la maille. Les opérations de symétrie les plus couramment appliquées aux cristaux de macromolécules biologiques sont les rotations, les translations et les « tour de de vis » ("screw axes", combinaisons [rotation - translation]). Cristallographie : assemblage biologique L'assemblage biologique (ou unité biologique - "biological assembly") est l'assemblage macromoléculaire qui est ou semble être la forme fonctionnelle de la molécule. Par exemple, la forme fonctionnelle de l'hémoglobine est constituée de 2 x 2 chaînes polypeptidiques (α2β2). Selon la structure cristalline, des opérations de symétrie peuvent être nécessaires pour obtenir l'assemblage biologique complet. A l'inverse, un sous-ensemble des coordonnées cristallographiques peut suffire pour représenter l'assemblage biologique. En conclusion, un assemblage biologique peut être construit à partir :
Exemples
Outils d'analyse des assemblages biologiques Des bases de données spécifiques, telles que PISA ("Protein Interfaces, Surfaces and Assemblies"), permettent d'analyser les assemblages biologiques de la PDB. jsPISA est un outil web interactif pour le calcul des surfaces macromoléculaires et des interfaces, pour l'évaluation de leurs propriétés et pour l'inférence d'assemblages macromoléculaires probables à partir de données de coordonnées (généralement cristallographiques). |
Cristallographie : : les fichiers au format mmCIF ("macromolecular Crystallographic Information Format") Un fichier au format mmCIF contient les instructions pour générer un assemblage biologique. Un grand nombre de programme de visualisation moléculaire (dont Jmol) sont compatibles avec ce format. Un fichier mmCIF contient donc les informations concernant les éléments structuraux qui générent un assemblage biologique. Ces informations se trouvent dans des catégories (ou listes d'informations) appelées "pdbx_struct_assembly", "pdbx_struct_assembly_gen" et "pdbx_struct_oper_list" :
La catégorie "struct_biol" contient les remarques spécifiques des auteurs relatives aux assemblages biologiques. Un dictionnaire de données archive les expériences de cristallographie de petites molécules et leurs résultats. Voir le fichier "mmcif_pdbx.dic" ("PDB Exchange Dictionary - PDBx/mmCIF"). Le format de ce dictionnaire et les fichiers de données basés sur ce dictionnaire sont conformes à la représentation des données appelée STAR ("Self Defining Text Archive and Retrieval"). Extrait d'un fichier au format mmCIF _pdbx_struct_assembly.id 1 _pdbx_struct_assembly.details author_and_software_defined_assembly _pdbx_struct_assembly.method_details PISA _pdbx_struct_assembly_gen.assembly_id 1 _pdbx_struct_assembly_gen.asym_id_list A,B,C,D,E,F,G,H loop_ _pdbx_struct_assembly_prop.biol_id 1 'ABSA (A^2)' 3840 ? loop_ _pdbx_struct_oper_list.id _pdbx_struct_oper_list.matrix[1][1] 1 'identity operation' 1_555 1.0000000000 0.0000000000 2 'crystal symmetry operation' 4_565 1.0000000000 0.0000000000 1_555 : décrit l'opérateur de symétrie utilisé (désigné par le nombre 1) et les opérations de translation nécessaires (le chiffre 555). Les opérateurs de symétrie sont définis par le groupe d'espace et les opérations de translation sont indiquées pour les 3 axes de la cellule unitaire (a, b et c) :
4_565 : utilisation de l'opérateur de symétrie 4, suivie d'une opération de translation de la cellule unitaire dans la direction positive selon l'axe b. |
b. La résonance magnétique nucléaire Voir le principe de la RMN. Du fait de son caractère non destructif, la RMN est employée en biologie et en chimie organique pour déterminer la structure de certaines protéines ou de fragments d'ADN, de molécules organiques, ... C'est une technique où les molécules sont en solution. L'un des avantages de la RMN est d'obtenir des informations sur la dynamique des arrangements conformationnels au sein des macromolécules biologiques par mesure des temps de relaxation (T1, T2), des temps de corrélation, des vitesses d'échange chimique. Ces arrangements conformationnels peuvent être :
Kurt Wüthrich a reçu le prix Nobel de chimie en 2002 pour le développement de la RMN pour la détermination de la structure des macromolécules en solution. Terminologie : a. Protéine non marquée par des isotopes ("2D Homonuclear nuclear magnetic resonance" - spectre RMN en 2 dimensions) : davantage appliqué aux peptides et aux petites protéines.
b. Attribution des déplacements chimiques observés à chaque type d'atome (marquage isotopique / carbone 13 et azote 15) : expérience HSQC ("2D Heteronuclear Single Quantum Correlation") pour les noyaux autres que l'hydrogène. c. Grosses protéines : expérience TROSY ("Transverse Relaxation Optimized SpectroscopY"). d. Protéines membranaires et fibrillaires : elles sont "sous-étudiées" structuralement car les agents chimiques chaotropes (détergents) nécessaires à leur solubilisation des membranes limitent (voire empêchent) l'obtention de cristaux qui diffractent. Une méthode est de plus en plus employée pour déterminer la structure de ces protéines : "Magic-angle spinning solid-state NMR (MAS ssNMR) spectroscopy". Voir : "Magic-Angle Spinning (MAS)". |
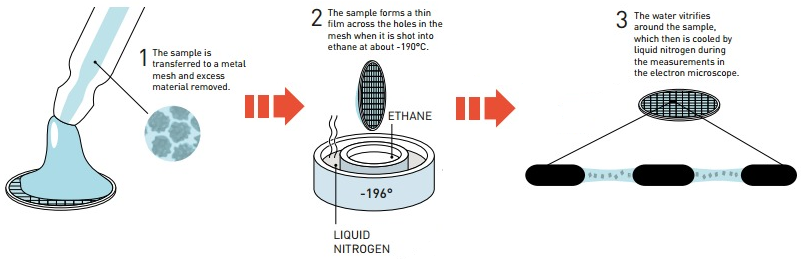
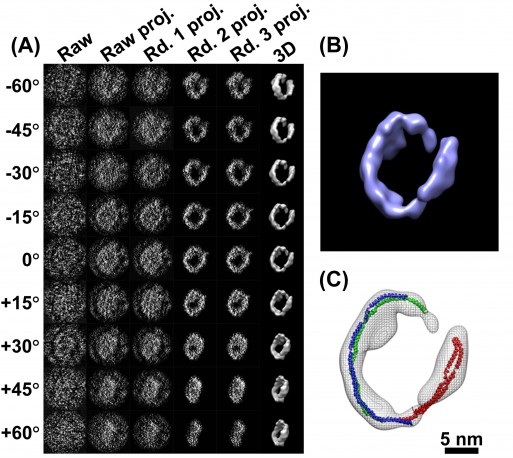
c. La cryo-microscopie électronique La cryo-microscopie électronique ("single-particle cryo-electron microscopy" - Cryo-EM) est une forme de microscopie électronique à transmission où l'échantillon est étudié à des températures cryogèniques (azote liquide, environ -195°C). Dans ces conditions de congélation rapide, l'eau n'a pas le temps de former des cristaux.
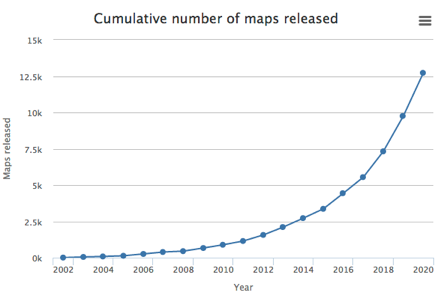
Source : Jarnestad J. - The Royal Swedish Academy of Sciences La cryo-EM permet donc l'observation d'échantillons dans leur état natif, non cristallin, par opposition à la diffraction des rayons X qui requière en général des conditions non physiologiques. Les besoins en quantité de matériel biologique purifié sont moindres que pour la diffraction des rayons X ou la RMN. Voir un article expliquant les principes de diverses techniques de préparation d'échantillons vitrifiés pour pour la cryo-EM : Weissenberger et al., 2021. EM Databank ("Unified Data Resource for 3-Dimensional Electron Microscopy") : Base de données de structures déterminées par cryo-EM. La cryo-EM est de plus en plus utilisée pour la détermination de la structure des macromolécules biologiques (ci-dessous, les statistiques de la base de données EMDB).
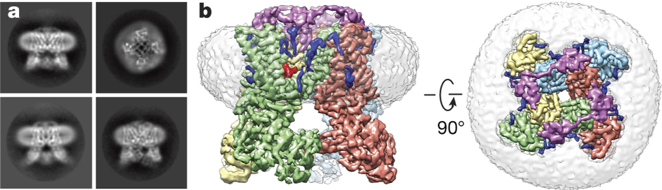
La cryo-EM est surtout utilisée pour les complexes biologiques (virus, ribosome, spliceosome, ...) : en effet, leur très grande taille empêche de les étudier par diffraction des rayons X ou RMN. La résolution de la cryo-EM est désormais comparable à celle des deux autres grandes techniques de détermination de la structure des macromolécules biologiques. Pour obtenir des modèles à l'échelle atomique, il est nécessaire d'affiner ("in silico modeling") les données de densité électronique de cryo-EM avec celles des structures cristallographiques d'entités constitutives de ces complexes. Figure ci-dessous : Evolution des technologies de Cryo-EM.
Source : Fujiyoshi, Y. (2011) Le microscope enregistre les données d'un trés grand nombre de particules orientées au hasard. Un modèle tri-dimensionnel est ensuite reconstruit par ordinateur à partir d'images 2D sélectionnées. Figure ci-dessous :
Source : Berkeley lab |
Les avancées de la cryo-microscopie électronique Plusieurs facteurs sont cause d'une perte importante d'information dans les images de cryo-EM (et donc de la limitation de la résolution actuelle de cette technique) :
L'utilisation de nouveaux détecteurs d'électrons dits "à conversion directe" ("direct-conversion electron detectors") et d'algorithmes de correction des mouvements spécifiquement développés à cet usage a démontré que cette technique permet de résoudre des structures macromoléculaires à une résolution quasi atomique. Ces caméras ont non seulement des performances améliorées dans la détection [signal-bruit], mais surtout elles sont suffisamment rapides pour suivre les mouvements de particules lors de l'irradiation par les électrons. Ainsi les mouvements de l'échantillon (pendant l'acquisition des données) induits par l'énergie du faisceau peuvent être corrigés. Deux exemples d'application :
Les protéines membranaires Lorsque les protéines membranaires intégrales sont visualisées dans des détergents ou d'autres systèmes artificiels, une information capitale est perdue : les interactions avec les lipides et leurs effets sur la structure réelle de la protéine dans la membrane. C'est d'autant plus marquant dans le cas des protéines pour lesquelles les lipides ont un rôle structural et un rôle de régulation. La cryo-EM / couplée à la technologie des lipides nanodisques permet de déterminer la structure d'une protéine membranaire dans une bicouche lipidique. Exemple : l'étude du récepteur ionotrope TRPV1 ("Transient Receptor Potential cation channel subfamily V member 1") du rat. Ce type de récepteur est activé par des molécules de la famille des vanilloïdes (exemple : la capsaïcine du piment) ou une température supérieure à 42°C.
Source : Gao et al. (2016) La cryo-EM / couplée à la technologie des lipides nanodisques :
|
Prix Nobel 2017 Le Prix Nobel de Chimie a été attribué en 2017 à Jacques Dubochet, Joachim Frank et Richard Henderson pour le développement de la technique de cryo-EM.
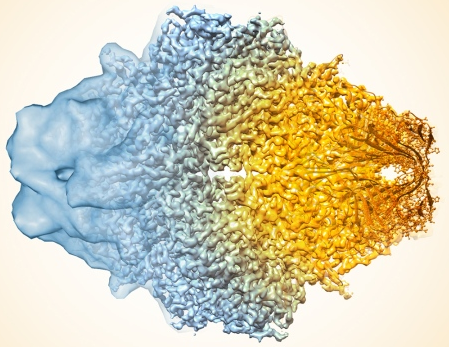
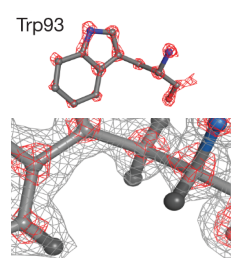
Source : Cressey & Callaway (2017) "Cryo-electron microscopy wins chemistry Nobel" La résolution de la cryo-EM a notablement augmenté en évoluant d'une carte de densité à basse résolution (partie gauche de la structure de la β-galactosidase, figure ci-dessous) aux coordonnées atomiques (environ 2 Å - partie droite). Augmentation remarquable de la résolution de la cryo-EM La structure de la ferritine (stockage du fer) à été déterminée avec une résolution d'environ 1,2 Å par reconstruction cryo-EM à particule unique : les données sont de qualité suffisante pour observer les atomes individuels dans l'apoferritine (en absence de fer). Cette amélioration remarquable de la résolution repose sur des progrès matériels (Yip et al., 2020 ; Nakane et al., 2020).
Source : Yip et al. (2020) |
d. Autres techniques moins fréquemment utilisées Ces techniques sont pour la plupart des techniques de pointe, d'avancée récente et ultra-sophistiquées. La diffusion des rayons X aux petits angles ("Small-angle X-ray scattering" - SAXS) :
La diffusion des neutrons aux petits angles ("Small angle neutron scattering" - SANS) :
La spectroscopie infrarouge par transformée de Fourier ("Fourier Transform Infrared Spectroscopy" - FTIR) :
La localisation en microscopie à force atomique ("Localization Atomic force microscopy" - LAFM) :
La fluorescence et les fluorochromes :
La spectromètrie de masse : étude de l'assemblage de protéines (pour l'instant jusqu'à 1 million Da) par spectromètrie de masse "Orbitrap mass analyser" (quadrupôle / "time of flight"). |
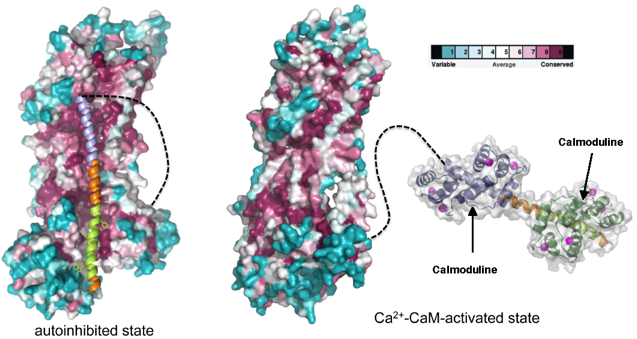
Exemple d'utilisation de plusieurs méthodes Modèle d'activation de la Ca2+-ATPase :
Figure ci-dessous - à gauche : forme auto-inhibée de la Ca2+- ATPase de la membrane plasmique ("Plasma-Membrane Ca2+-ATPase"). Figure ci-dessous - à droite : fixation de deux molécules de calmoduline (complexée au calcium) sur les sites de fixation à haute affinité (en vert clair et en bleu clair). Cette fixation déplace l'hélice auto-inhibitrice du coeur catalytique, ce qui active la pompe à ion.
Source : Tidow et al. (2012) |
4. La mécanique et la modélisation moléculaires Ce type d'approche est complémentaire des techniques physiques qui précèdent. Ces objectifs sont entre autres :
Voir le principe de la mécanique moléculaire et la notion de champs de force. Exemple de terminologie anglo-saxonne : "Backbone torsion angles optimization with Monte Carlo minimization protocol" / "Energy minimization using a quasi-Newton method" / "Lazaridis–Karplus implicit solvation model". Différents outils informatiques sont utilisés pour :
Voir une liste quasi exhaustive des programmes de mécanique et modélisation moléculaires. |
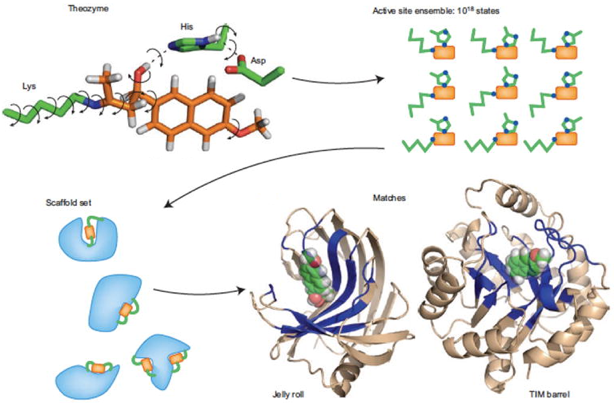
5. Les méthodes "ab initio" ("depuis le commencement") Il y a un grand nombre, fini, de repliements des protéines observés dans la nature. A ce jour on comptabilise environ 1400 repliements (selon les modes de classification et les bases de données). On ne sait pas si les structures non encore observées sont physiquement impossibles ou si elles n'ont pas encore été "testées" par le processus évolutif ou caractérisées par les biologistes structuraux. Les méthodes informatiques (algorithmiques) de conception de nouvelles structures protéiques :
sont un moyen de répondre (peut-être) rapidement à cette question mais aussi de concevoir des protéines "artificielles" aux propriétés thérapeutiques originales. Exemples de champs d'application :
b. Démarche "Template-based modeling" Le but est de générer une séquence ou un ensemble de séquences d'acides aminés qui se replie(nt) dans une structure 3D préalablement déterminée ("template").
Limitations actuelles
|
c. Description schématique de la démarche "Template-based modeling" 1ère étape Génération des séquences d'acides aminés susceptibles de déboucher sur un repliement donné ("template"). Ci-dessous : cette équation décrit un moyen de générer ces séquences d'acides aminés "artificielles".
Source : Fung et al. (2008) 1er cas : une structure protéique de départ ("template")
La fonction à minimiser est la somme des énergies d'interactions 2 à 2 entre les acides aminés de la séquence de départ. Le terme Ejlik (xi, xk) est l'énergie d'interaction entre la position i occupée par l'acide aminé j et la position k occupée par l'acide aminé l. Ce terme dépend :
2ème cas : plusieurs structures protéiques de départ Le terme Ejlik (xi, xk) est remplacé par un terme d'énergie moyenne pondéré : La distance entre xi etxk est donc remplacée par une distance moyenne pondérée entre toutes les structures. Des centaines, voire des milliers de séquences potentielles ("decoys") peuvent ainsi être générées. Deuxième étape Recherche des conformations les plus stables thermodynamiquement (fonctions de minimisation d'énergie) et semblables à la structure native :
La stabilité des modèles ("decoys") est évaluée par des fonctions de scores qui combinent :
Des étapes finales d'affinements pour augmenter la résolution de la structure native calculée sont parfois nécessaires. Voir la procédure suivie par "ASTRO-FOLD". Troisième étape Les conformations proches ou équivalentes à la structure native ("native-like conformation") sont alors sélectionnées : (i) sur la base de ces fonctions de score; (ii) par regroupement de conformères semblables. |
d. Démarche "de novo protein design" - Rosetta L'originalité de cette méthode est qu'elle ne s'appuie sur aucune structure 3D préalablement déterminée. Terminologies équivalentes : "de novo structure prediction" / "de novo structure modeling". Rosetta est un projet dédié à la prédiction de nouvelles structures de protéines ("ab initio protein structure prediction method Rosetta") par calculs partagés sur ordinateurs ou grille de calcul distribué (plateforme BOINC - "Berkeley Open Infrastructure for Network Computing").
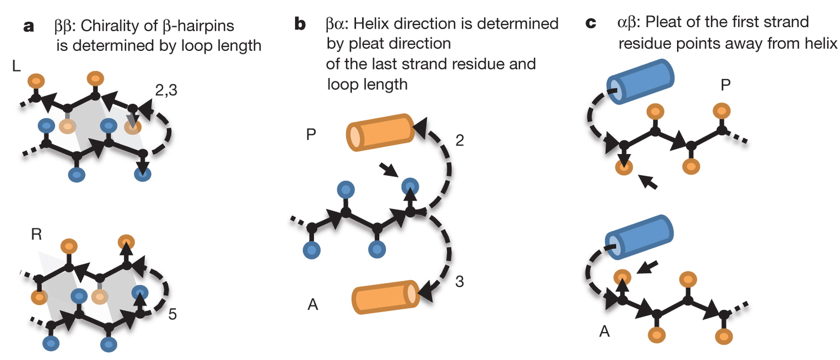
Source : Nanda & Koder (2010) Des règles trés précises de topographie des acides aminés au sein de structures secondaires ont pu être énoncées (Koga et al., 2012) :
Source : Koga et al. (2012) Aller à la base de données "Motivated proteins" : elle contient un trés grand nombre de motifs stabilisés par des liaisons hydrogène et des règles qui en découlent. Ci-dessous, aperçu de quelques méthodes de modélisation et de cadres de conception de macromolécules dans l'environnement Rosetta.
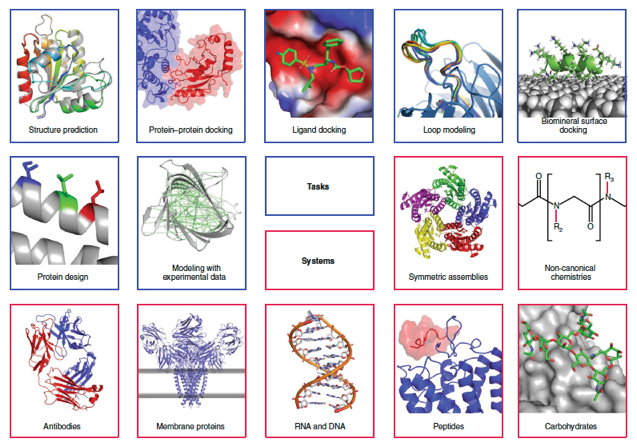
Source : Leman et al. (2020) |
Les protéines naturelles ont évolué pour reconnaître un ensemble relativement faible de molécules de ligand avec une grande affinité et une grande spécificité. Élargir cet ensemble de couples [protéine - ligand] avec des protéines synthétiques spécifiques de ces ligands pourrait modifier radicalement le développement de biocapteurs, de médicaments à base de protéines, d'enzymes artificielles et autres outils pour la biologie chimique. De nouvelles méthodes de calcul utilisent la sélection virtuelle du meilleur conformère dans un très vaste ensemble de conformations (Tinberg et al., 2013).
Exemple de lignes de commande pour générer les conformères: Exemple de lignes de commande pour la recherche de structures concordantes : Exemple de lignes de commande pour le "design" de structures: |
e. Exemple de la protéine "artificielle" TOP7 (2003) C'est une protéine "artificielle" de 93 acides aminés issue de simulations / calculs de prédiction ("de novo protein design") effectués par Brian Kuhlman et Gautam Dantas (équipe de David Baker - Université de Caroline du Nord) . Ces chercheurs ont utilisé comme point de départ un repliement encore jamais mis en évidence dans la nature. Les séquences ont été générées avec le programme "Rosetta design Monte Carlo search protocol and energy function" : (i) un potentiel de Lennard-Jones 12-6; (ii) un terme pour les liaisons hydrogène dépendant de l'orientation; (iii) un modèle de solvatation implicite. Tous les acides aminés (excepté la cystéine) ont été autorisés pour 71 des 93 positions (≈ 110 rotamères par position) et les 22 positions restantes (surface des feuillets) ont été restreintes à des acides aminés polaires (≈ 75 rotamères par position). L'espace de recherche était de 11071 × 7522, soit ≈ 10186 rotamères. Les conformations du squelette carboné ont été générées sans contrainte pour optimiser la compacité des chaînes latérales : en conséquence, les séquences de plus basse énergie avaient une énergie très supérieure à celle de protéines natives de même taille.
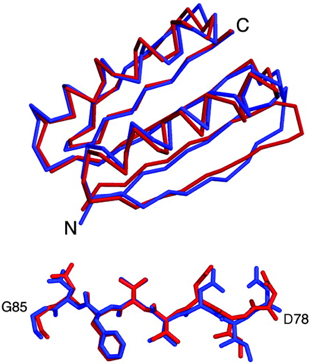
La comparaison de la structure modèle conçue par calcul et de la structure cristalline est remaquable (figure ci-dessous) :
Source : Kuhlman et al. (2003) Ensemble de la chaîne carbonée : RMSD = 1,17 Å - Peptide [Asp78 - Gly85] : RMSD = 0,79 Å |
|
Visualisation de TOP7 à une résolution de 2,5 Å Le chargement de la structure peut prendre du temps. Code PDB : 1QYS
|
| 6. Liens Internet et références bibliographiques |
|
PDB : Protein Data Bank SCOP : Structural Classification of Proteins TOPS : Topology of Protein Structure database CASP : Critical Assessment of Techniques for Protein Structure Prediction |
|
|
Rosetta@home Foldit : jeu en ligne de prédiction de structures de protéines basé sur la plate-forme Rosetta. Folding@home The AlphaFold Protein Structure Database (DeepMind EMBL-EBI) : accès ouvert aux prévisions de la structure des protéines pour le protéome humain et d'autres organismes clés. |
|
|
CAMEO ("Continuous Automated Model EvaluatiOn") : projet communautaire visant à évaluer en permanence la précision et la fiabilité des serveurs de prédiction de la structure des protéines de manière entièrement automatisée. The Protein Model Portal : module de PSI-KB ("Protein Structure Initiative Knowledgebase") |
|
|
HPF : Human Proteome Folding Project TOP7 : molecule of the month - PDB SBKB : PSI Structural Genomics Knowledgebase (PSI : Protein Structure Initiative) Motivated proteins : A Web Facility for Studying Small Hydrogen-Bonded Motifs (très beau travail pédagogique) |
|
|
GROMACS : a versatile package to perform molecular dynamics, i.e. simulate the Newtonian equations of motion for systems with hundreds to millions of particles I-TASSER : "Protein structure and function predictions" GalaxyWEB : "Web server for protein structure prediction and refinement" |
|
|
Kuhlman et al. (2003) "Design of a Novel Globular Protein Fold with Atomic-Level Accuracy" Science 302, 1364 - 1368 Fung et al. (2008) "Toward Full-Sequence De Novo Protein Design with Flexible Templates for Human Beta-Defensin-2" Biophys J. 94, 584 - 599 Nanda & Koder (2010) "Designing Artificial Enzymes by Intuition and Computation" Nat. Chem. 2, 15 - 24 Fujiyoshi, Y. (2011) "Structural physiology based on electron crystallography" Protein Sci. 20, 806 - 817 |
|
|
Koga et al. (2012) "Principles for designing ideal protein structures" Nature 491, 222 - 227 Tidow et al. (2012) "A bimodular mechanism of calcium control in eukaryotes" Nature 491, 468 - 472 Rose et al. (2012) "High-sensitivity Orbitrap mass analysis of intact macromolecular assemblies" Nat. Meth. 9, 1084 - 1086 Vogeli et al. (2012) "Spatial elucidation of motion in proteins by ensemble-based structure calculation using exact NOEs" Nat. Struc. Mol. Biol. 19, 1053 - 1057 |
|
|
Shahid et al. (2012) "Membrane-protein structure determination by solid-state NMR spectroscopy of microcrystals" Nature Meth. 9, 1212 - 1217 Gopinath & Veglia (2012) "Dual Acquisition Magic-Angle Spinning Solid-State NMR-Spectroscopy: Simultaneous Acquisition of Multidimensional Spectra of Biomacromolecules" Angew Chem. Int. Ed. Engl. 51, 2731 - 2735 Banigan & Traaseth (2012) "Utilizing Afterglow Magnetization from Cross-Polarization Magic-Angle-Spinning Solid-State NMR Spectroscopy to Obtain Simultaneous Heteronuclear Multidimensional Spectra" J. Phys. Chem. B 116, 7138 - 7144 Inokuma et al. (2013) "X-ray analysis on the nanogram to microgram scale using porous complexes" Nature 495, 461 - 466 |
|
|
Bai et al. (2013) "Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles" eLife 2, e00461 Li et al. (2013) "Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM" Nat. Methods 10, 584 - 590 Tinberg et al. (2013) "Computational design of ligand-binding proteins with high affinity and selectivity" Nature 501, 212 - 216 Gao et al. (2016) "TRPV1 structures in nanodiscs reveal mechanisms of ligand and lipid action" Nature 534, 347 - 351 |
|
|
Le Prix Nobel de Chimie a été attribué en 2017 à Jacques Dubochet, Joachim Frank et Richard Henderson pour le développement de la technique de cryomicroscopie électronique. Henderson et al. (1990) "Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy" J. Mol. Biol. 213, 899 - 929 Dubochet, J. (2016) "A reminiscence about early times of vitreous water in electron cryomicroscopy" Biophys. J. 110, 756 - 757 Cressey & Callaway (2017) "Cryo-electron microscopy wins chemistry Nobel" Nature 550, 167 |
|
|
Leman et al. (2020) "Macromolecular modeling and design in Rosetta: recent methods and frameworks" Nat. Methods 17, 665 - 680 Yip et al. (2020) "Atomic-resolution protein structure determination by cryo-EM" Nature Nakane et al. (2020) "Single-particle cryo-EM at atomic resolution" Nature |
|
|
Weissenberger et al. (2021) "Understanding the invisible hands of sample preparation for cryo-EM" Nat. Methods 18, 463 - 471 Jumper et al. (2021) "Highly accurate protein structure prediction with AlphaFold" Nature 596, 583 - 589 Baek et al. (2021) "Accurate prediction of protein structures and interactions using a three-track neural network" Science 373, 871 - 876 |
![]()