
| La protéomique |
| Tweet |
|
|
1. Introduction 2. L'extraction des protéines 3. L'électrophorèse sur gel bi-dimensionnel
4. La spectrométrie de masse 5. Principe de l'analyse protéomique par spectrométrie de masse |
6. Quelques données pour l'interprétation des spectres de masse de peptides
7. Identification des protéines par des méthodes bioinformatiques
8. Protéomique quantitative 9. Exemples d'application 10. Liens Internet et références bibliographiques |
|
1. Introduction La protéomique a pour but d'identifier (et de quantifier) l'ensemble des protéines synthétisées ou protéome, à un moment donné et dans des conditions données au sein d'un tissu, d'une cellule ou d'un compartiment cellulaire. Le protéome est extrêmemement complexe à plusieurs titres :
La transcriptomique analyse l'ensemble des transcrits (produits d'expression des gènes). La protéomique et la transcriptomique sont donc des approches complémentaires trés puissantes qui peuvent être utilisées pour des études fondamentales ou appliquées en biologie, en médecine, en agriculture, ... En effet, dans les deux cas, les infomations recueillies permettent d'aborder l'ensemble des réponses cellulaires dans leur globalité et non plus de manière partielle. La protéomique apporte des réponses auxquelles la transcriptomique ne peut répondre :
Les études de protéomique sont de plus en plus spécialisées :
Le taux de croissance annuel composé du marché mondial de la protéomique* est estimé à 14,2 % pour atteindre 17,2 milliards de dollars en 2017. *Technologies liées à la protéomique : puces à protéines ("protein microarray"), spectrométrie de masse et spectroscopie RMN, fractionnement des protéines, chromatographie, électrophorèse, "Surface Plasmon Resonance", cristallographie et diffraction des rayons X, immuno-essais ... |
| Génome et protéome de l'homme | ||
| Analyse aboutie du génome de l'homme | 2012 | "The ENCODE Project Consortium" :
20 687 gènes codant des protéines Nature 489, 57-74 (2012) |
| Protéome de l'homme | 2014 | Kim et al. (2014) "A draft map of the human proteome" Nature 509, 575-581 |
|
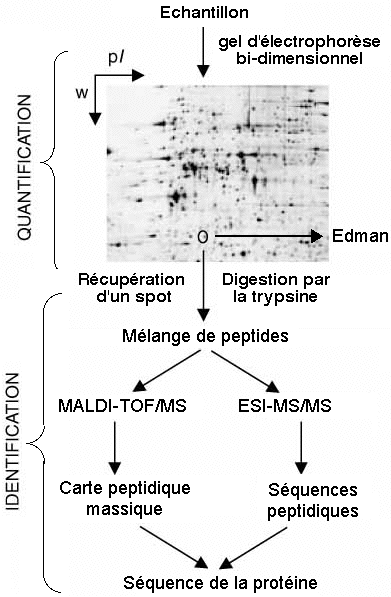
Schématiquement, les étapes de l'analyse protéomique sont les suivantes :
Figure adaptée de Peng & Gygi (2001)
|
|
C'est une étape cruciale car elle peut entraîner une dégradation ou une dénaturation irréversible des protéines et compromettre leur identification. Pour la solubilisation des protéines hydrosolubles, on utilise :
Les protéines membranaires, constituées de beaucoup d'acides aminés hydrophobes, sont plus difficiles à solubiliser. Leur extraction peut être améliorée en utilisant :
L'analyse du protéome, non plus de la cellule entière, mais de compartiments subcellulaires permet d'étudier des protéines trés spécifiques ou en faible quantité du fait d'un faible taux de synthèse. Exemple : l'analyse protéomique de la mitochondrie du pois (Bardel et al., 2002). C'est ainsi qu'une équipe de l'Université d'Angers (IRHS) a été la première à isoler et à caractériser une protéine LEA mitochondriale (Grelet et al., 2005). Voir la base de données "Late Embryogenesis Abundant Proteins Database" (Hunault, Châtelain & Jaspard) |
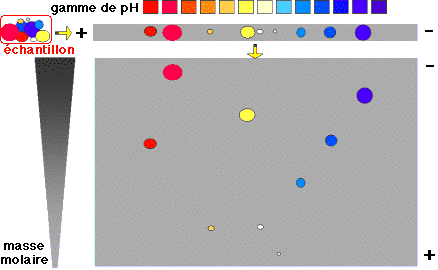
| 3. L'électrophorèse sur gel bi-dimensionnel (SWISS-2DPAGE) La stratégie la plus courante est de séparer dans un premier temps les protéines par électrophorèse bidimensionnelle sur un gel de polyacrylamide (ou 2D - PAGE - "two-dimensional polyacrylamide gel electrophoresis"). Du fait de leur composition en acides aminés, les protéines diffèrent les unes des autres par leur masse molaire et leur charge nette à un pH donné. La charge nette est caractérisée par le point isoélectrique ou pI. Les protéines ont :
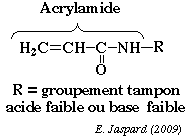
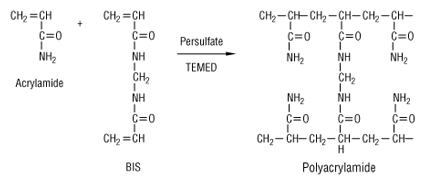
L'une des difficultés majeures de la séparation est de trouver des conditions d'électrophorèse qui couvrent de telles gammes de propriétés physico-chimiques. La séparation des protéines extraites se fait en deux temps. 1ère dimension : une isoélectrofocalisation (IEF) où les polypeptides migrent jusqu'à un pH égal à leur pl. La séparation selon la charge utilisait auparavant des ampholytes pour obtenir un gradient de pH. Mais ce procédé n'était pas assez reproductible pour la comparaison de gels par superposition. On utilise des gradients de pH immobilisés (IPG), formés avec des immobilines. Les immobilines sont des dérivés de l'acrylamide et leur structure générale est décrite dans la figure ci-desous.
R correspond à un groupement carboxyle (acide) ou à une amine tertiaire (base) qui forment une série de molécules tampon avec différentes valeurs de pKa d'ionisation (exemples : 3,6 - 4,4 - 4,6 - 6,2 - 7,0 - 8,5 - 9,3). Les immobilines co-polymérisent avec les molécules d'acrylamide (voir figure ci-dessus) dans le gel d'IEF. Ainsi, les groupes chargés portés par les immobilines qui forment le gradient de pH sont covalamment attachés (via une liaison vinyle) à la matrice de polyacrylamide et donc immobilisés. Cette technique permet d'obtenir des gradients de pH reproductibles et très étroits : une pente de 0.01 unité pH / cm peut séparer un mélange de protéines avec une différence de pI de 0.001 unité pH. 2ème dimension : une séparation selon la masse molaire sur un gel d'électrophorèse de polyacrylamide en conditions dénaturantes (sodium dodécyl sulfate ou SDS et réduction des ponts disulfure).
Source : Piercenet Ces deux électrophorèses sont effectuées de manière perpendiculaire l'une par rapport à l'autre.
Source : Humboldt - Universität Berlin Comme les paramètres de la séparation sont indépendants, cette technique est particulièrement résolutive : plusieurs centaines de chaînes polypeptidiques peuvent être séparées sous forme de taches de protéines (appelées aussi spots) sur un gel. Voir des exemples de gels bi-dimensionnels. |
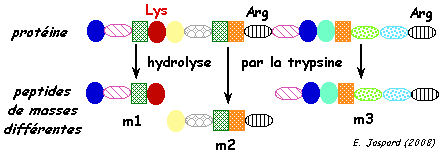
| c. L'hydrolyse des protéines par la trypsine La ou les protéines contenue(s) dans un spots de l'électrophorèse bi-dimensionnelles est (sont) hydrolysée(s) en fragments peptidiques par une protéase à sérine (essentiellement endopeptidase). En général, on utilise la trypsine (EC 3.4.21.4), la LysC, la chymotrypsine. La trypsine hydrolyse la liaison peptidique après (côté C-terminal) les acides aminés lysine et arginine sauf si ces acides aminés sont suivis par une proline. Cependant elle peut avoir une spécificité plus complexe. Ces peptides ont des masses différentes car leur séquence en acides aminés sont différentes.
1. Voir le logiciel "PeptideCutter" - Expasy 2. Aller à la page "MS-Digest" du logiciel "Prospector". Cliquer sur "perform digest" pour avoir un aperçu d'un résultat d'hydrolyse in silico d'une protéine. |
|
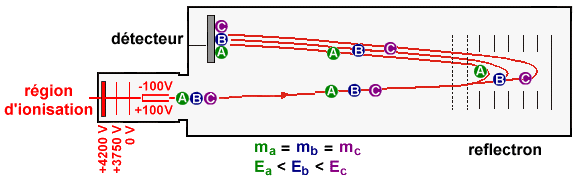
4. La spectrométrie de masse L'ionisation électronique (souvent appelée impact électronique) et l'ionisation chimique sont les principales méthodes d'ionisation. Dans le cas de l'ionisation électronique, l'échantillon est introduit dans une enceinte sous vide, il y est vaporisé puis soumis au bombardement d'un canon à électrons de grande énergies. Un électron est arraché aux molécules et on obtient une espèce qui est à la fois un cation (ion positif) et un radical libre (nombre impair d'électrons), que l'on appelle ion moléculaire, M+.: M + e- (énergie 70 eV) <===> M+. + 2 e- L'énergie du faisceau ionisant fragmente l'ion moléculaire par rupture des liaisons les plus faibles avant les liaisons les plus fortes et donne naissance à des ions positifs de masses plus faibles, qui pourront être fragmentés à nouveau (exemple : spectrométrie de masse dite en tandem - MS/MS). Ces ions sont ensuite accélérés dans un champ électrique et/ou magnétique, puis dirigés entre les pôles d'un aimant selon une trajectoire circulaire qui dépend de leur rapport masse/charge [m/z]. En faisant varier le champ électrique, on fait varier la vitesse des ions moléculaires et on peut les faire ainsi parvenir au détecteur par ordre croissant de rapport [m/z]. Le tri des ions s'effectue :
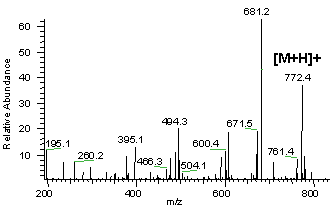
Remarque : z étant pratiquement toujours égal à 1, on obtient une mesure de la masse de tous les fragments et de la molécule initiale [M+H]+. On obtient un grand nombre de pics, tous de masse inférieure à celle de l'ion moléculaire. Cet ensemble constitue un diagramme de fragmentation. Les groupements fonctionnels possèdent un diagramme de fragmentation qui leur sont propres. Dans un spectre de masse, la hauteur relative des pics indique l'abondance relative des espèces.
Source : "Ion source" |
|
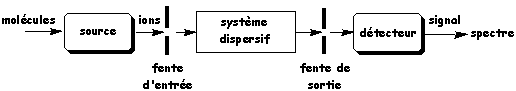
b. Description d'un spectromètre de masse Un spectromètre de masse est constitué de cinq parties principales (figure ci-dessous) :
Source : IUT Lannion 1. Le système qui introduit l'échantillon dans le spectromètre de masse. Par exemple un système de chromatographie en phase gazeuse ("gaz chromatography" - GC). 2. La source où à lieu l'ionisation des molécules et la fragmentation des ions. Il existe plusieurs méthodes d'ionisation, le choix de celle-ci est directement lié à la nature de l'échantillon et au type d'analyse souhaitée :
Pour l'analyse des protéines, deux méthodes sont principalement utilisées : la désorption par effet de champ ou MALDI et l'électrospray ou ESI. 3. Le système dispersif ou analyseur qui sépare et trie les ions suivant leur rapport [m/z]. Cette partie diffère d'un appareil à l'autre car il existe plusieurs modes d'ionisation selon les applications. 4. Le détecteur qui collecte les ions (fragments) qui arrivent à des temps différents en fonction du rapport [m/z] et qui amplifie le signal associé aux ions. Pour l'analyse des protéines, les détecteurs sont : la trappe à ions (IT), le temps de vol (TOF), le quadripôle (MS ou Q). 5. L'ensemble informatique de traitement des données qui permet de transformer les informations reçues par le détecteur en spectre de masse. Les spectromètres de masse actuels permettent d'enregistrer automatiquent plusieurs milliers de spectres MS/MS en quelques heures. L'interprétation de ces spectres de masse et l'identification des protéines (analyse bioinformatique) sont le facteur limitant. |
| 5. Principe de l'analyse protéomique par spectrométrie de masse Synoptique de l'analyse par spectromètrie de masse
Source : Vandenbrouck et al. (2005) |
|
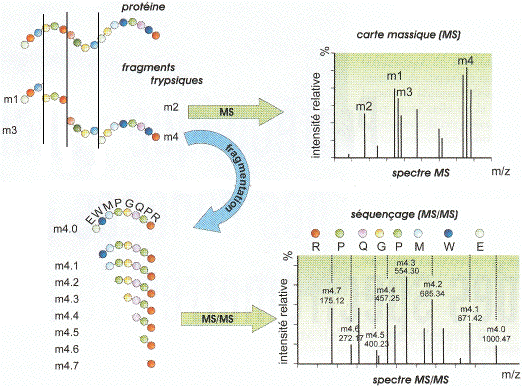
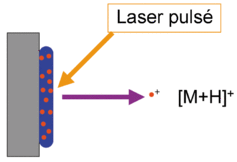
a. La carte peptidique massique par spectromètrie de masse : MALDI-TOF Le type de spectromètrie de masse utilisé est l'ionisation par désorption au laser d'une matrice - temps de vol ("Matrix-Assisted Laser Desorption/Ionisation-Time of Fligh" - MALDI-TOF). Karas & Hillenkamp (1988) "Laser desorption ionization of proteins with molecular mass exceeding 10 000 Daltons" Anal. Chem. 60, 2299 - 2301 Le phénomène d'ionisation utilise une matrice cristallisée. Exemple : un mélange acide sinapinique / acétonitrile / H2O (50:50:0,1). Les propriétés de ce mélange sont :
La protéine est donc hydrolysée par la trypsine et les peptides qui en résultent sont vaporisées puis dispersées dans la matrice.
Source : Wikipédia - bleu : matrice - rouge : échantillon Sous l'effet d'un laser UV (337 nm), la matrice irradiée subit une désorption entraînant une ionisation des peptides qu'elle contient par transfert de proton. Dans les réflecteurs à temps de vol ("Time of Fligh" -TOF), les ions acquièrent une grande énergie cinétique et sont séparés en fonction de leur vitesse. Le temps de vol est fonction du rapport [m/z] de chaque peptide.
Source : "Introduction à la spectromètrie de masse" - P. Dubreuil Cette technique de spectrométrie de masse permet d'établir la carte peptidique massique ("peptide mass fingerprint - PMF") de la protéine ou empreinte. Henzel et al. (2003) "Protein identification: the origins of peptide mass fingerprinting" J. Am. Soc. Mass. Spectrom. 14, 931-942 |
|
b. Le séquençage par spectromètrie de masse en tandem : ESI-MS/MS ou ESI-Q-TOF
Quand on a un mélange de protéines trop complexe au sein d'un spot, l'identification d'une protéine en particulier est impossible car plusieurs empreintes se superposent dans la carte peptidique massique expérimentale issue de l'analyse par MALDI-TOF. Il faut alors séquencer les fragments peptidiques provenant de l'hydrolyse par la trypsine. On emploie la spectrométrie de masse en tandem ou MS/MS, où chacun des peptides issus de la première fragmentation est lui-même fragmenté.
Source : Li & Assmann (2000) Les produits de fragmentation sont alors des acides aminés ou des sous-peptides trés courts. La séquence complète de la protéine est reconstituée par chevauchement des séquences des différents peptides. Le type de spectromètrie de masse le plus couramment utilisé pour le séquençage est l'ionisation par électronébuliseur ("ElectroSpray Ionisation" - ESI-MS/MS). Les fragments peptidiques issus de l'hydrolyse par la trypsine sont en solution aqueuse volatile (mélange méthanol-eau 1:1). Cette solution est introduite dans la source par un capillaire trés fin chargé et en métal (figure ci-dessous).
Source : Cech & Enke (2002) L'ionisation des molécules a lieu dans des conditions douces (pression atmosphérique et température ambiante) : sous l'effet d'un gaz nébuliseur (N2) et d'un champ électrique, les gouttes "explosent" (la fission Coulombique) en un brouillard de gouttelettes chargée (un électrospray). Des ions multi-chargés comme [M + 2H]2+ sont souvent observés. La technique ESI-MS/MS :
Caractéristiques communes aux techniques d'ionisation MALDI (à partir d'une phase solide) et ESI (à partir d'une phase liquide) :
Exemple d'appareillage :
|
|
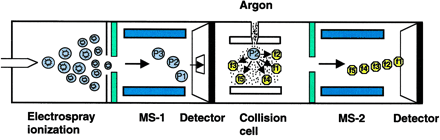
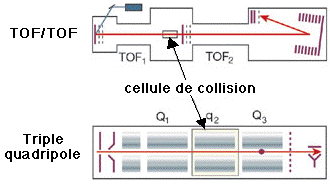
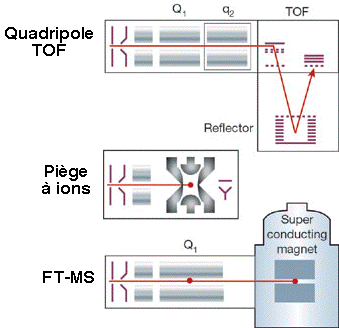
c. Autres types de spectromètres de masse 1. Dans les réflecteurs TOF/TOF (figure ci-dessous), une cellule de collision est située entre les les deux sections TOF. Les ions caractérisés par un certain rapport [m/z] sont séléctionnés dans la première section TOF puis fragmentés dans la cellule de collision. Les fragments sont séparés dans la seconde section TOF. 2. Une combinaison fréquemment rencontrée est de type [source électrospray - triple analyseur quadripôle] (figure ci-dessous) :
3. Les spectromètres de masse à quadripôles/TOF combinent le premier quadripôle (Q1), la cellule de collision (q2) et un réflecteur TOF (figure ci-dessous).
Source des figures ci-dessus : Aebersold & Mann (2003) "Mass spectrometry-based proteomics" Nature 422, 198 - 207 4. Le piège à ion 3D capture les ions comme dans le cas du piège à ion linéaire et il fragmente les ions caractérisés par un certain rapport [m/z]. Puis les fragments sont analysés (spectre de masse en tandem). 5. Les spectromètres de masse à transformée de Fourier ("Fourier transform ion cyclotron resonance mass spectrometer" - FT-ICR/MS) capturent les ions mais à l'aide d'un fort champs magnétique (5 à 10 Tesla). La figure montre la combinaison avec un piège à ions linéaire pour une plus grande efficacité d'isolement, de fragmentation et de détection des fragments. |
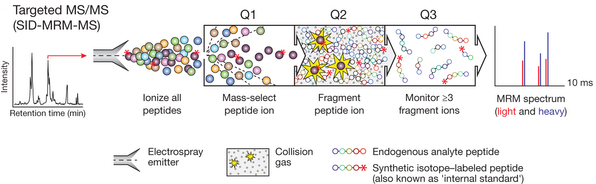
La protéomique ciblée ("Targeted mass spectrometry") Elle repose sur une technique de spectrométrie de masse appelée "suivi de réaction sélectionnée" ("selected reaction monitoring" - SRM). Principe : à l'aide d'un spectre de masse de référence, un analyte peut être identifié sur la base de quelques ions pré-sélectionnés sans nécessiter l'intégralité (complexe) du spectre de fragmentation MS/MS. Les données de SRM sont obtenues en réglant les deux analyseurs de masse d'un spectromètre de masse à triple quadripôle de façon qu'ils ne stabilisent que les trajectoires d'ions avec des valeurs [m/z] prédéfinies. Le filtrage des masses à 2 niveaux augmente la sélectivité et cette technique permet d'obtenir un rapport signal/bruit élevé pour les analytes cibles. Des peptides de synthèse contenant des étiquettes avec des isotopes stables servent de standard internes (quantités connues d'isotopes) et permettent une quantification précise d'un analyte. Un spectromètre de masse à triple quadripôle est constitué :
Source : Gillette & Carr (2013)
Le quadripôle Q1 est réglé pour sélectionner un ion de masse connue, qui est fragmenté dans le quadripôle Q2. Le quadripôle Q3 est réglé pour balayer la gamme entière de [m/z], donnant des informations sur les tailles des fragments générés. |
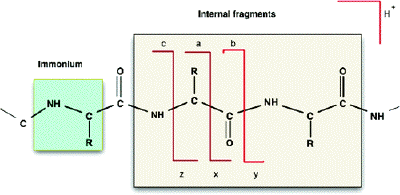
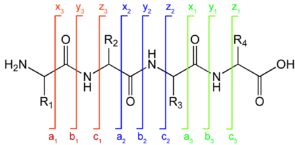
6. Quelques données pour l'interprétation des spectres de masse de peptides a. Nomenclature des fragments peptidiques issus de la spectromètrie de masse en tandem
Les ions (sous-peptides ou acides aminés) qui résultent de la fragmentation des peptides par ionisation / collision sont notés de la manière suivante :
Source : Khatun et al. (2007)
1. Type d'ions obtenus si la fragmentation a lieu au niveau de la liaison peptidique :
2. Type d'ions obtenus si la fragmentation a lieu au niveau d'une autre liaison :
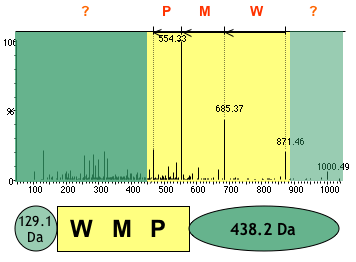
3. ion immonium : ion composé d'un fragment interne d'un seul résidu d'acide aminé (clivage mixte de type a et de type y). Figure ci-dessous : diagramme de fragmentation d'un peptide.
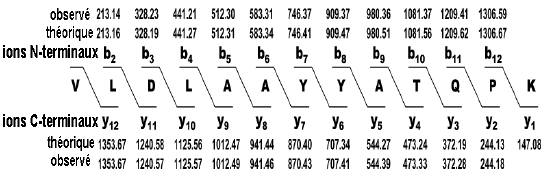
Figure ci-dessous : séquence du peptide déduite du diagramme de fragmentation. Les valeurs de masses des ions b ou y sont celles indiquées sur le diagramme ci-dessus.
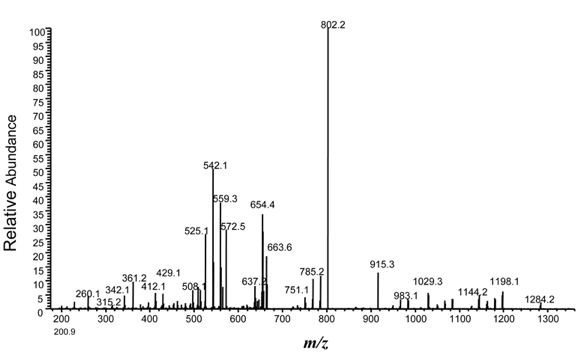
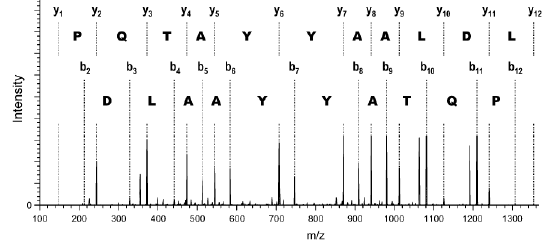
Source des figures : Suh et al. (2011) Figures ci-dessous : spectre de masse et type d'ions générés pour le peptide VLDLAAYYATQPK. Le chiffre qui accompagne la lettre du type d'ion indique le nombre d'acides aminés dans le fragment.
Source : Branca et al. (2007) |
|
b. Valeurs de masses monoisotopiques et moyennes des résidus d'acides aminés |
| Acide aminé (code une lettre) | Valeurs de masse monoisotopique | Moyenne | Acide aminé (code une lettre) | Valeurs de masse monoisotopique | Moyenne |
| Alanine (A) | 71.03711 | 71.0788 | Leucine (L) | 113.08406 | 113.1594 |
| Arginine (R) | 156.10111 | 156.1875 | Lysine (K) | 128.09496 | 128.1741 |
| Asparagine (N) | 114.04293 | 114.1038 | Methionine (M) | 131.04049 | 131.1926 |
| Acide aspartique (D) | 115.02694 | 115.0886 | Phénylalanine (F) | 147.06841 | 147.1766 |
| Cystéine (C) | 103.00919 | 103.1388 | Proline (P) | 97.05276 | 97.1167 |
| Acide glutamique (E) | 129.04259 | 129.1155 | Serine (S) | 87.03203 | 87.0782 |
| Glutamine (Q) | 128.05858 | 128.1307 | Thréonine (T) | 101.04768 | 101.1051 |
| Glycine (G) | 57.02146 | 57.0519 | Tryptophan (W) | 186.07931 | 186.2132 |
| Histidine (H) | 137.05891 | 137.1411 | Tyrosine (Y) | 163.06333 | 163.1760 |
| Isoleucine (I) | 113.08406 | 113.1594 | Valine (V) | 99.06841 | 99.1326 |
| Voir le tableau qui permet de connaitre la différence de masse induite par une substitution (valeurs de masse monoisotopique) - (ExPASy - "Protein Identification Tools") |
| Modification post-traductionnelle | Différence de masse (monoisotopique / moyenne) | Modification post-traductionnelle | Différence de masse (monoisotopique / moyenne) |
| Acétylation | 42.0106 / 42.0373 | Méthylation | 14.0157 / 14.0269 |
| ADP-ribosylation | 541.0610 / 541.30 | Myristoylation | 210.1984 / 210.3598 |
| Amidation | -0.9840 / -0.9847 | Palmitoylation | 238.2297 / 238.4136 |
| Biotinylation | 226.0776 / 226.2934 | Phosphorylation | 79.9663 / 79.9799 |
| Hydroxylation | 15.9949 / 15.9994 | Sulfatation | 79.9568 / 80.0642 |
|
Quelques règles pour calculer la masse d'un peptide :
Pour un cours plus détaillé sur l'interprétation des spectres de masse en protéomique, aller à "De Novo Peptide Sequencing Tutorial" - "IonSource.Com". |
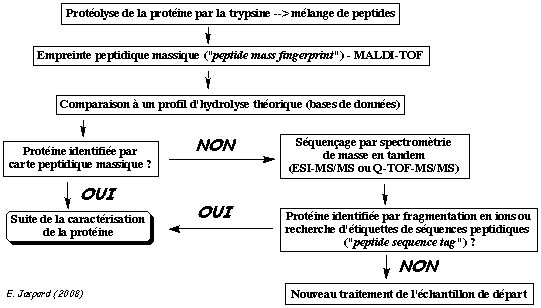
| 7. Identification des
protéines par des méthodes bioinformatiques
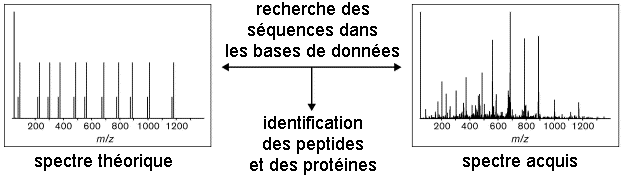
La bioinformatique permet d'analyser les spectres MS/MS pour identifier les protéines d'un échantillon complexe. Deux voies d'identification se distinguent par le type de banque utilisée pour le criblage : banque de séquences protéiques vs. banque de séquences génomiques. Dans le cas des banques de séquences protéiques, on distingue deux approches dites "directe" et "indirecte". Elle correspond à l'identification automatique des protéines par une analyse MS/MS virtuelle à partir de toutes les séquences protéiques d'une banque de données. Les séquences des protéines d'une banque de données sont hydrolysées in silico puis pour chaque peptide résultant, un spectre MS/MS théorique est calculé : on obtient une empreinte ou PMF théorique.
Voir : Chamrad et al. (2004) Exemple de logiciel : "Prospector", "Mascot", "Profound" Ensuite, le spectre de la protéine inconnue est comparé à l'ensemble de ces spectres théoriques.
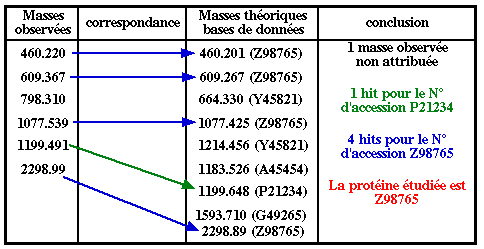
L'approche directe est basée sur l'information de masse des peptides : elle est donc rapide. L'approche directe a deux limites :
Remarque : La tolérance massique ("mass tolerance") est un paramètre important des logiciels (100 ppm = 1000.0 ± 0.1 Da). |
|
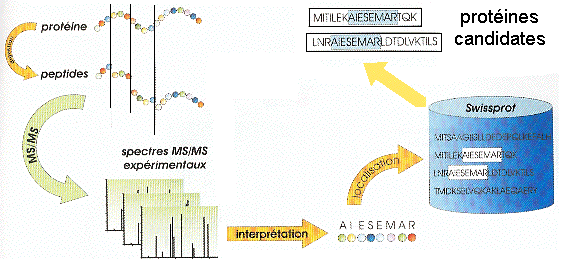
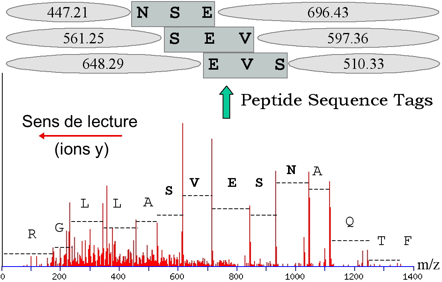
L'identification de la protéine étudiée en passe par une étape intermédiaire : l'interprétation du spectre MS/MS qui consiste à le convertir en une ou plusieurs séquence(s) peptidique(s) possible(s). C'est ce que l'on appelle le séquençage "de novo". Cette ou ces séquence(s) potentielle(s) est/sont ensuite comparée(s) à celles des banques de données comme SwissProt par exemple. L'approche indirecte s'appuient donc sur des logiciels qui recherchent les similarités entre séquences. Or ces logiciels sont conçus pour accepter des variations (substitution, insertion ou délétion d'acides aminés). Ainsi, l'approche indirecte (contrairement à l'approche directe) permet de repérer dans les banques de données des séquences en acides aminés non totalement identiques mais présentant des analogies avec la séquence déduite du décryptage du spectre MS/MS.
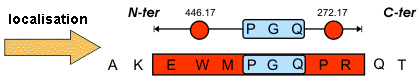
Source : Vandenbrouck et al. (2005) La limitation de l'approche indirecte réside dans l'interprétation du spectre MS/MS qui est coûteuse en temps. Comme certaines régions des spectres MS/MS sont difficiles à interpréter, Matthias Mann et Matthias Wilm ont préconisé de restreindre l'interprétation de ces spectres à la détermination d'une sous-séquence que l'on appelle étiquette peptidique ("Peptide Sequence Tags" - PST). Une PST est donc (figure ci-dessous) :
Source : "Bioinformatique en protéomique"
Ces étiquettes sont utilisées pour cribler les banques de données. |
|
c. PepLine : annotation des génomes par l'analyse MS/MS Les logiciels "Taggor", "PMMatch" et "PMClust" (création Rhône-Alpes-Génopole / "Helix" - INRIA Rhône-Alpes / Genome Express) constituent la suite logiciel "PepLine" qui permet une annotation automatisée des génomes à partir des données de spectromètrie de masse MS/MS. 1. Etape de conversion / interprétation des spectres MS/MS : le module "Taggor" génère toutes les étiquettes (séquence de 3 acides aminés) possibles à partir d'un spectre MS/MS et leur attribue un score. Les 10 (en général) meilleures PST sont retenues.
Source : CEA 2. Etape de localisation : le module "PMMatch" localise les PST fournis par le module "Taggor" sur les séquences des banques (protéines ou ADN génomique).
Source : Vandenbrouck et al. (2005)
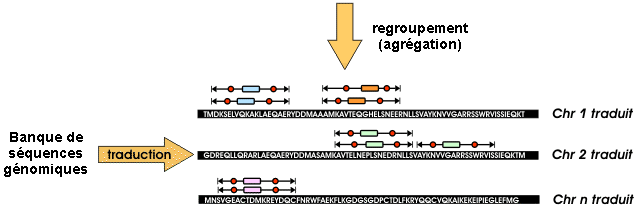
3. Etape de regroupement : le module "PMClust" regroupe ("cluster") les hits trouvés.
Source : Vandenbrouck et al. (2005) Le regroupement des hits permet d'associer les PST à une protéine ou à un gène. La validité statistique du regroupement est évaluée par un score.
|
| Quelques programmes pour l'analyse bioinformatique en protéomique | |
| Recherche dans les bases de données | Mascot |
| SEQUEST | |
| InSpecT | |
| PEAKS | |
| Tandem | |
| Sipros | |
| séquençage "de novo" | PEAKS |
| PepNovo | |
| Lutefisk | |
| Recherche d'homologie d'étiquettes ("Tag homology search") | CIDentify |
| SPIDER | |
| MS-BLAST | |
| FASTA | |
| MS-Homology | |
| GutenTag | |
| Séquençage de protéines | CHAMPS |
| CSPS | |
| "Cross-linked peptides" | xQuest / xProphet |
| pLink | |
|
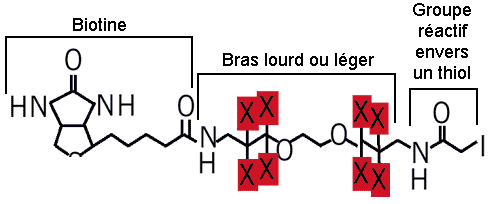
8. Protéomique quantitative La technique de marqueurs d'affinité contenant un isotope d'identification ("Isotope Coded Affinity Tags" - ICAT) permet de comparer l'abondance relative des protéines entre 2 échantillons (exemple : condition normale vs. condition pathologique). Le groupement thiol -SH des cystéines des protéines réagit avec l'iodoacétamide des réactifs ICAT. Les réactifs ICAT possèdent 8 atomes d 'hydrogène portés par un bras espaceur (figure ci-dessous) :
Source : Gygi et al. (1999)
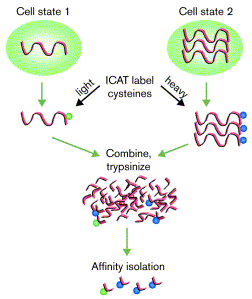
Chaque échantillon est marqué par un seul type de réactif. Les 2 échantillons sont mélangés et les protéines de ce mélange sont hydrolysées par la trypsine. Les peptides sont séparés par chromatographie d'affinité sur avidine par l'intermédiaire du groupement biotine porté par les réactifs.
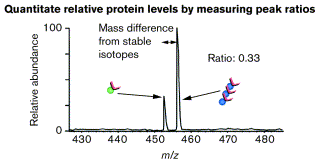
Source : Gygi et al. (2000) Les peptides identiques sont élués ensemble puisqu'ils ne différent que par la présence de l'isotope. Les peptides identiques sont analysés ensemble par spectromètrie de masse, ce qui permet de quantifier leur abondance relative et d'identifier les protéines.
Source : Gygi et al. (2000) |
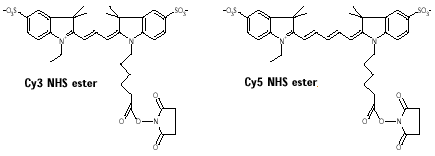
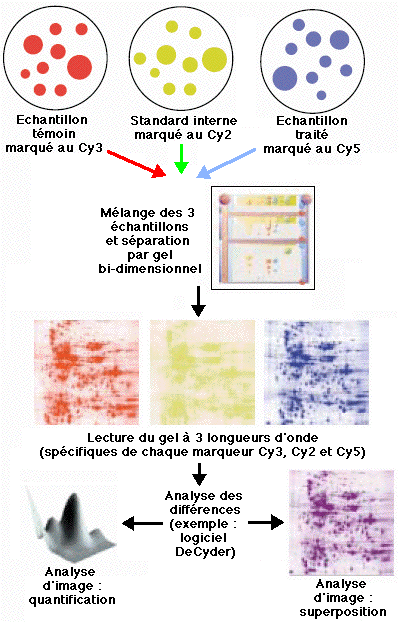
b. La technique 2D-DIGE ("two-dimensional fluorescence DIfference Gel Electrophoresis") Unlu et al. (1997) "Difference gel electrophoresis: a single gel method for detecting changes in protein extracts" Electrophoresis 18, 2071 - 2077 Cette technique est basée sur le marquage direct des lysines (réaction du groupement ε-aminé avec le groupement ester du pigment) des protéines de 3 échantillons distincts par des pigments fluorophore cyanine :
Source : GE Healthcare
Les 3 échantillons sont mélangés et une électrophorèse bi-dimensionnelle est effectuée sur un même gel.
Source : MedicalProteomics A la différence de la technique ICAT, la quantification repose sur l'analyse d'images (exemple : "Typhoon™") et non sur une comparaison des quantités relatives de protéines entre les échantillons. Celà souligne l'importance d'un standard interne pour les étapes ultérieures de normalisation (exemple : logiciel "DeCyder™"). |
|
c. Protéomique "en vrac" ("Shotgun Proteomics") α. "Multidimensional Protein Identification Technology" - MudPIT - Washburn et al. (2001) Principe de MudPIT :
La technologie MudPIT est un processus hautement automatisé qui permet la caractérisation de milliers de protéines et, en particulier, de protéines :
C'est cependant une démarche trés onéreuse et trés gourmande en moyen d'analyse informatique. Par ailleurs, le problème majeur de cette approche "en vrac" est l'ambiguité pour assigner des peptides communs à différentes protéines, à l'inverse de l'approche par gel bi-dimensionnel où les protéines sont séparées au préalable. β. Les techniques de chromatographies pour l'étude de peptides spécifiques De nombreuses études de protéomique focalisent sur des familles spécifiques de peptides. Les peptides issus de l'hydrolyse enzymatique des protéines de l'échantillon sont séparés et enrichis par des techniques de chromatographie liquide à haute performance ("high Performance Liquid Chromatography" - HPLC) avant l'analyse par spectromètrie de masse. Cette approche est fréquemment utilisée pour l'étude de profils d'expression quantitatifs car elle est supposée introduire un biais quantitatif moindre dans l'échantillon biologique. Chromatographie des phospho-peptides: un moyen pour enrichir le contenu en peptides phosphorylés d'un échantillon est la chromatographie d'affinité sur métaux immobilisés ("Immobilized Metal Affinity Chromatography" - IMAC). Un ion métallique (fer ou gallium) est fixé à une résine d'acide imino-diacétique. Ce complexe de cation trivalent fixent les phospho-peptides (et les peptides fortement acides) par chélation. Hydrophobicité: c'est un paramètre physico-chimique très important de certains peptides utilisé en chromatographie d'hydrophobicité ("Ion-pairing Reversed-Phase High-Performance Liquid Chromatography" - RP-HPLC). La chromatographie d'affinité : des protéines ou des complexes protéiques spécifiques, des protéines de la surface cellulaire sont sélectivement capturés par un ligand immobilisé sur la résine. Par exemple, des anticorps pour l'immunoprécipitation, des petites molécules pour le marquage des sites actifs, le complexe [lectine/hydrazide] pour la capture de protéines glycosylées, ... L'isolement des peptides N-terminaux par la technique COFRADIC ("COmbined FRActional DIagonal Chromatography") : 2 séparations chromatographiques consécutives identiques avec une étape de modification chimique entre les 2 séparations qui permet de cibler le sous-ensemble des peptides N-terminaux. La chromatographie d'échange d'ions : des techniques combinées de chromatographie d'échange d'anions puis de cations permettent d'analyser des peptides de faible abondance (Zhou et al., 2010). γ. Depôts publics de données, consortium, bases de données en protéomique consortium "ProteomeXchange" |
|
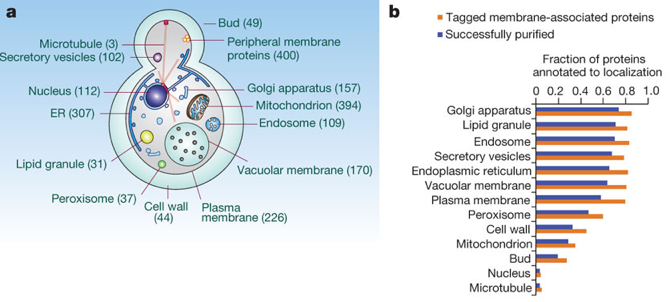
a. Le protéome membranaire de la levure Les assemblages macromoléculaires impliquant des protéines membranaires ont des rôles biologiques vitaux et sont des cibles privilégiées de médicaments. Une étude protéomique a été effectuée pour purifier à grande échelle ("large-scale affinity purification") puis caractériser le plus grand nombre de complexes [protéines - protéines membranaires] de Saccharomyces cerevisiae. Ces complexes ont été solubilisés à partir des divers types de membranes des organites par l'utilisation de détergents non-dénaturants. Des complexes impliquant 1.144 protéines membranaires intégrales (intrinsèques), 400 protéines membranaires périphériques (extrinsèques) et 46 protéines membranaires ancrées dans la membrane via des lipides ont ainsi été isolées. L'identité des protéines co-purifiées a été déterminée par spectromètrie de masse en tandem et une annotation en fonction des compartiments cellulaires (Gene Ontology).
Source : Babu et al. (2012)
Cette étude a permis d'établir une carte très précise de 1.726 interactions physiques entre [protéines co-purifiées et protéines membranaires] et de mettre en évidence 501 complexes hétéromèriques associés aux divers systèmes membranaires. |
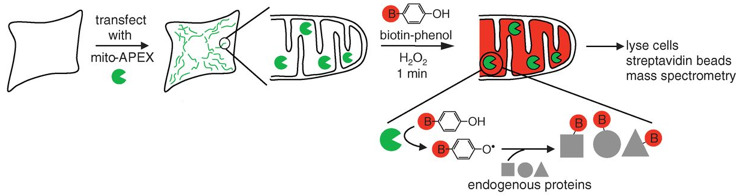
b. Le protéome de la matrice mitochondriale de l'homme Les méthodes traditionnelles pour les études protéomiques d'organites se traduisent souvent par une spécificité limitée des protéines étudiées, la perte de matériel biologique et d'éventuelles contaminations du fait des étapes d'isolement des organites et de purification de leur contenu en protéines. Une méthode récente permet de minimiser ces inconvénients : elle utilise un marquage spécifique des protéines de la matrice de la mitochondrie par la biotine, tandis que la cellule est vivante, avec toutes ses membranes et ses complexes protéiques intacts et que les relations spatiales entre les protéines sont préservées. La méthode utilise une enzyme, l'ascorbate peroxidase (APEX), modifiée par ingénierie. L'APEX est spécifiquement adressée à la matrice mitochondriale par fusion à un peptide d'adressage de 24 acides aminés. Une fois dans la matrice mitochondriale, l'APEX marque par biotinylation (liaison covalente) les protéines voisines (mais pas les protéines lointaines) dans les cellules vivantes. L'APEX est active dans tous les compartiments cellulaires et elle oxyde de nombreux dérivés du phénol en radicaux phénoxyl. Ces radicaux :
Le marquage covalent est fait par l'addition de [biotine (B) - phénol] et de H2O2. Les cellules sont ensuite lysées et les protéines biotinylées sont récupérées avec des billes sur lesquelles est fixée la streptavidine. Les protéines sont ensuite éluées, séparées sur gel et identifiées par spectromètrie de masse.
Source : Rhee et al. (2013) Cette étude a permis :
Source : Rhee et al. (2013) |
| 10. Liens Internet et références bibliographiques |
|
SWISS-2DPAGE Two-dimensional polyacrylamide gel electrophoresis database (ExPASy) ExPASy Proteomics tools Vandenbrouck et al. (2005) "Protéomique : analyse des données issues des spectromètres de masse" Biofutur 252, 29 -31 |
|
|
"Mt PROTEOMICS : a site dedicated to the symbiotic proteomics of Medicago truncatula root" (INRA - CNRS - Université de Bourgogne) "The Plant Proteome Database for Arabidopsis thaliana and Zea mays" "Arabidopsis thaliana Seed Proteome" (CNRS - INRA - Bayer) |
|
|
Agrawal et al. (2010) "Plant secretome: Unlocking secrets of the secreted proteins" Proteomics 10, 799 - 827 Zhou et al. (2010) "Analysis of low-abundance proteins using the proteomic reactor with pH fractionation" Talanta 80, 1526 -1531 Lancaster et al. (2011) "A computational framework for proteome-wide pursuit and prediction of metalloproteins using ICP-MS and MS/MS data" BMC Bioinformatics 12, 64 Kristensen et al. (2012) "A high-throughput approach for measuring temporal changes in the interactome" Nat. Methods 9, 907 -909 Dix et al. (2012) "Functional Interplay between Caspase Cleavage and Phosphorylation Sculpts the Apoptotic Proteome" Cell 150, 426 - 440 Maarten Altelaar et al. (2013) "Next-generation proteomics: towards an integrative view of proteome dynamics" Nature Rev. Genet. 14, 35 - 48 |
|
|
Peng & Gygi (2001) "Proteomics: the move to mixtures" J. Mass Spectrom. 36, 1083 - 1091 Bardel et al. (2002) "A survey of the plant mitochondrial proteome in relation to development" Proteomics 2, 880 - 898 Grelet et al. (2005) "Identification in Pea Seed Mitochondria of a Late-Embryogenesis Abundant Protein Able to Protect Enzymes from Drying" Plant Physiol. 137, 157 - 67 |
|
|
Li & Assmann (2000) "Mass Spectrometry. An Essential Tool in Proteome Analysis" Plant Physiol. 123, 807 - 810 Cech & Enke (2002) "Practical implications of some recent studies in electrospray ionization fundamentals" Mass Spectrometry Reviews 20, 362 - 387 Khatun et al. (2007) "Fragmentation Characteristics of Collision-Induced Dissociation in MALDI TOF/TOF Mass Spectrometry" Anal. Chem. 79, 3032 -3040 Suh et al. (2011) "Using chemical derivatization and mass spectrometric analysis to characterize the post-translationally modified Staphylococcus aureus surface protein G" Biochim. Biophys. Acta 1804, 1394 - 1404 Gillette & Carr (2013) "Quantitative analysis of peptides and proteins in biomedicine by targeted mass spectrometry" Nature Methods 10, 28 - 34 |
|
|
Mann & Wilm (1994) "Error-tolerant identification of peptides in sequence databases by peptide sequence tags" Anal. Chem. 66, 4390 - 4399 Chamrad et al. (2004) "Evaluation of algorithms for protein identification from sequence databases using mass spectrometry data" Proteomics 4, 619 - 628 Branca et al. (2007) "De novo sequencing of a 21-kDa cytochrome c4 from Thiocapsa roseopersicina by nanoelectrospray ionization ion-trap and Fourier-transform ion-cyclotron resonance mass spectrometry" J. Mass Spectrometry 42, 1569 - 1582 Babu et al. (2012) "Interaction landscape of membrane-protein complexes in Saccharomyces cerevisiae" Nature 489, 585 - 589 |
|
|
Karas & Hillenkamp (1988) "Laser desorption ionization of proteins with molecular mass exceeding 10 000 Daltons" Anal. Chem. 60, 2299 - 2301 Fenn et al. (1989) "Electrospray ionization for mass spectrometry of large biomolecules" Science 246, 64 - 71 Unlu et al. (1997) "Difference gel electrophoresis: a single gel method for detecting changes in protein extracts" Electrophoresis 18, 2071 - 2077 Gygi et al. (1999) "Quantitative analysis of complex protein mixtures using isotope-coded affinity tags" Nature Biotechnol. 17, 994 - 999 Gygi et al. (2000) "Measuring gene expression by quantitative proteome analysis" Curr. Op. Biotech. 11, 396 - 401 Ferro et al. "Pepline: a software pipeline for high-throughput direct mapping of tandem mass spectrometry data on genomic sequences" J. Proteome Res. |
|
|
"Protein/peptide database ProMEX" Hummel et al. (2007) "ProMEX: a mass spectral reference database for proteins and protein phosphorylation sites" BMC Bioinformatics 8, 216 "AMPDB : the Arabidopsis Mitochondrial Protein Database" Heazlewood & Millar (2005) "AMPDB: the Arabidopsis Mitochondrial Protein Database" Nucleic Acids Res., 33, D605 - D610 La Société Française d'Electrophorèse et d'Analyse Protéomique (SFEAP) |
|
|
"La bioinformatique en protéomique : analyse des spectres de masse" - F. Rechenmann & I. Quinkal Site "Ion source" : spectromètrie de masse. Contient aussi des cours et exercices appliqués à la protéomique à faire en ligne. Master Protéomique - Université Lille |
|
|
Washburn et al. (2001) "Large-scale analysis of the yeast proteome by multidimensional protein identification technology" Nat. Biotechnol. 19, 242 - 247 Nesvizhskii & Aebersold (2005) "Interpretation of Shotgun Proteomic Data. The Protein Inference Problem" Molec. & Cell. Proteomics 4, 1419 - 1440 Roepstorff & Fohlman (1984) "Proposal for a common nomenclature for sequence ions in mass spectra of peptides" Biomed. Mass Spectrom.11, 601 Aebersold & Mann (2003) "Mass spectrometry-based proteomics" Nature 422, 198 - 207 Rhee et al. (2013) "Proteomic Mapping of Mitochondria in Living Cells via Spatially Restricted Enzymatic Tagging" Science 339, 1328 - 1331 |
|
![]()


{kind=link}