| L'annotation en génomique fonctionnelle et en protéomique |
| Tweet |
|
|
1. Rappel des buts de la génomique et de la protéomique 2. L'annotation : présentation générale 3. L'annotation des gènes avec l'ontologie 4. Les problèmes de l'annotation |
5. Les moyens pour l'annotation 6. Quelques méthodes et outils pour l'annotation 7. Liens Internet et références bibliographiques |
|
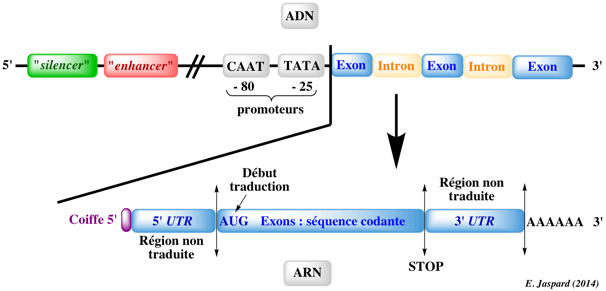
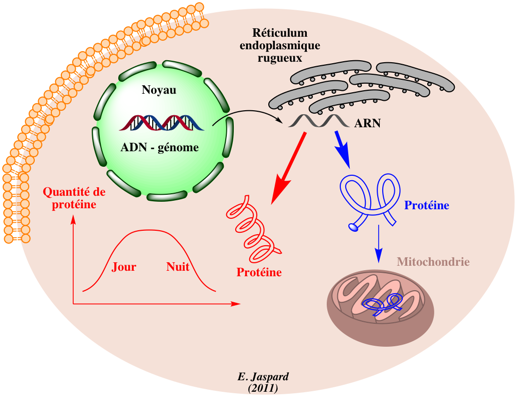
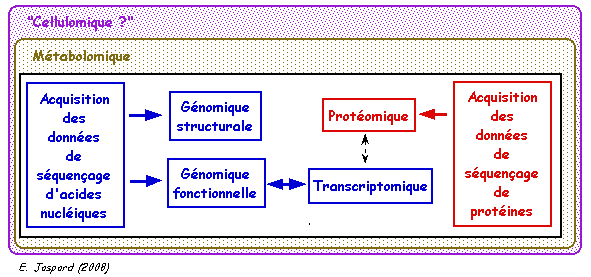
1. Rappel des buts de la génomique, de la transcriptomique et de la protéomique La génomique structurale analyse la structure des gènes et autres parties du génome. La génomique fonctionnelle analyse la fonction des gènes et autres parties du génome. Elle inclue l'analyse du transcriptome (ARN messagers) ou transcriptomique. Ces disciplines permettent l'annotation des génomes et l'identification des séquences informatives (les gènes avec ou sans introns codant des protéines ou des ARN, les séquences régulatrices, les séquences répétées, les éléments transposables, ...).
La protéomique a pour but d'identifier (et de quantifier) l'ensemble des protéines synthétisées ou protéome, à un moment donné et dans des conditions données au sein d'un tissu, d'une cellule ou d'un compartiment cellulaire. Le protéome est extrêmemement complexe à plusieurs titres :
|
La protéomique apporte des réponses auxquelles la transcriptomique ne peut répondre :
Les buts de ces disciplines sont donc (liste non exhaustive) :
|
|
2. L'annotation : présentation générale L’annotation d’un génome, d'un transcriptome d'un protéome, d'un métabolome ... consiste à documenter de la manière la plus exhaustive les informations issues de ces disciplines. On conçoit que c'est un travail encyclopédique de titan, d'autant que de nouvelles données s'accumulent qui doivent être croisées. L'adéquation entre fouille de données textuelles ("text mining") automatique pertinente et recherche "manuelle" par les experts fait l'objet de "workshops" internationaux ("BioCreative Article Classification Task" - BBCIII-ACT) et de nombreuses publications. a. L’annotation automatique s'appuie (essentiellement) sur des comparaisons des séquences à annoter avec les séquences présentes dans les banques de données. Les algorithmes recherchent des similarités / homologies de séquence, de structure, de motifs, … Ils permettent de prédire la fonction d’une molécule et de transfèrer automatiquement l'annotation entre les molécules homologues. Si l'annotation des molécules de référence est correcte, il n'y a pas de souci. Si elle est fausse, c'est un "jeu de domino" : l'erreur initiale est répercutée de proche en proche. b. L’annotation manuelle (ou curation) par des experts (des curateurs) qui valident ou invalident la prédiction en fonction de leurs connaissances ou de résultats expérimentaux. L'annotation manuelle est donc tout à fait indispensable. Mais, vue la quantité "astronomique" de données acquises quotidiennement, il est illusoire d'envisager une curation manuelle de l'ensemble des données en temps réel. On mesure aisément le problème : une quantité "minime" de données traitées par l'homme en temps réel qui induit un retard / décalage de plus en plus grand. c. L’annotation structurale dans le cas d'un génome tente de prédire :
Il existe des méthodes intrinsèques ou ab-initio qui s'appuient sur techniques informatiques d'apprentissage automatique utilisant :
Il existe des méthodes extrinsèques qui reposent sur la comparaison des ORF avec les séquences présentes dans les banques de données (exemples : "Orpheus", "Critica", "Reganor", ...). d. L’annotation fonctionnelle tente de prédire la fonction potentielle des gènes (notion d'étiquette, avec nom, fonction et interactions probables). e. L’annotation relationnelle tente de décrire les relations (interactions) entre les produits des gènes (familles de gènes, réseaux de régulation, réseaux métaboliques, ...). |
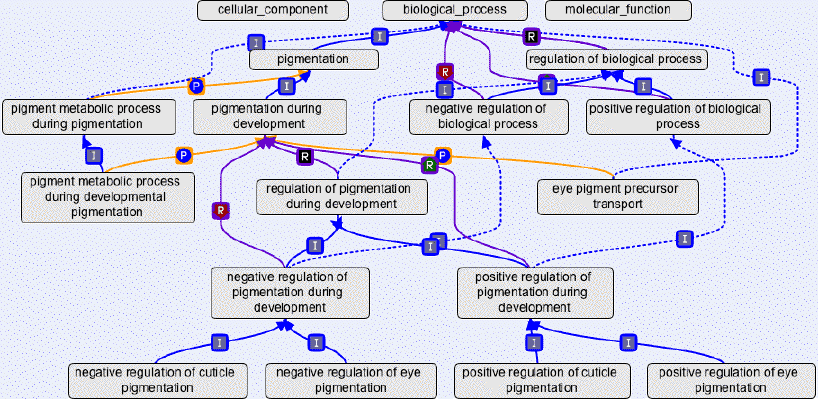
3. L'annotation des gènes avec l'ontologie a. L'ontologie et le consortium Gene Ontology Une ontologie est un ensemble structuré de termes et de concepts qui représentent le sens d'un champ d'informations, que ce soit par :
Chaque mot-clé de l'ontologie est associé à des lexicons (synonymes, homonymes, hyperonymes, ...). Le réseau autour d'un mot-clé est appelé concept. Les concepts sont formalisés sous forme d'un graphe au sein duquel il existe des relations sémantiques ou d'inclusion ("appartient à"). De manière schématique, on peut considérer qu'en génomique, l'ontologie est associée aux notions de terminologies et de classifications. Le consortium Gene Ontology (GO) :
|
|
Exemple : tous les termes suivants
|
décrivent la formation du glucose. |
D'où le terme GO : "gluconeogenesis" |
b. Les termes GO Le produit d'un gène :
Les termes GO sont les noeuds de l'ontologie. Il en existe actuellement plus de 32000 dans GO. GO décrit donc les produits des gènes via un ensemble de termes d'un graphe dirigé acyclique ("Directed Acyclic Graph" - DAG) qui contient 3 axes hiérarchiquement indépendants (CC, BP et MF, définis ci-dessus).
Source : GO Un terme GO est défini par :
|
| Résultat de GO en cherchant le terme "glycolysis" (ou, à l'inverse le code GO "0006096") | |
| Accession | 0006096 |
| Ontology | Biological Process |
| Synonyms |
|
| Definition | "The chemical reactions and pathways resulting in the breakdown of a monosaccharide (generally glucose) into pyruvate, ..." Source: GOC:bf, ISBN:0716720094, Wikipedia:Glycolysis |
| Subset | Prokaryotic GO subset |
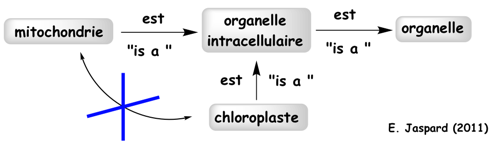
Les termes GO sont liés par un ensemble de relations : "Is a" - "Part of" - "Regulates" - "Positively Regulates" - "Negatively Regulates" - "Occurs in"
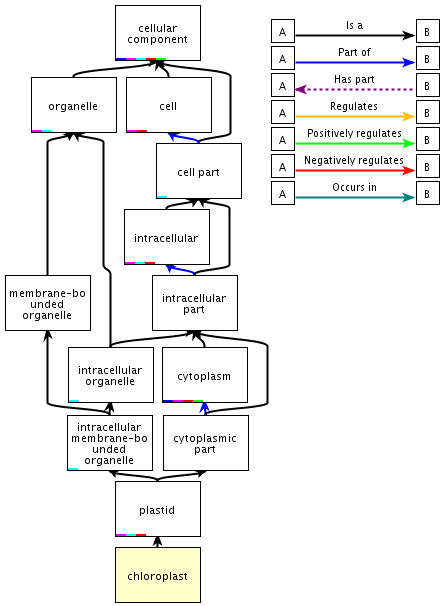
Voir un développement sur les relations de l'ontologie de GO. Chaque terme hérite de la signification de tous les termes qui le séparent de la racine de l'ontologie (notion d'ancêtre, parent et enfant). Ces termes sont donc placés dans une hiérarchie rigoureuse qui établit entre eux des liens univoques de "parents à enfants". Figure ci-dessous : illustration des liens entre les termes pour le mot "chloroplast" lui-même inclu dans le concept "cellular component".
Source : Annotation de la base de données "QuickGO" |
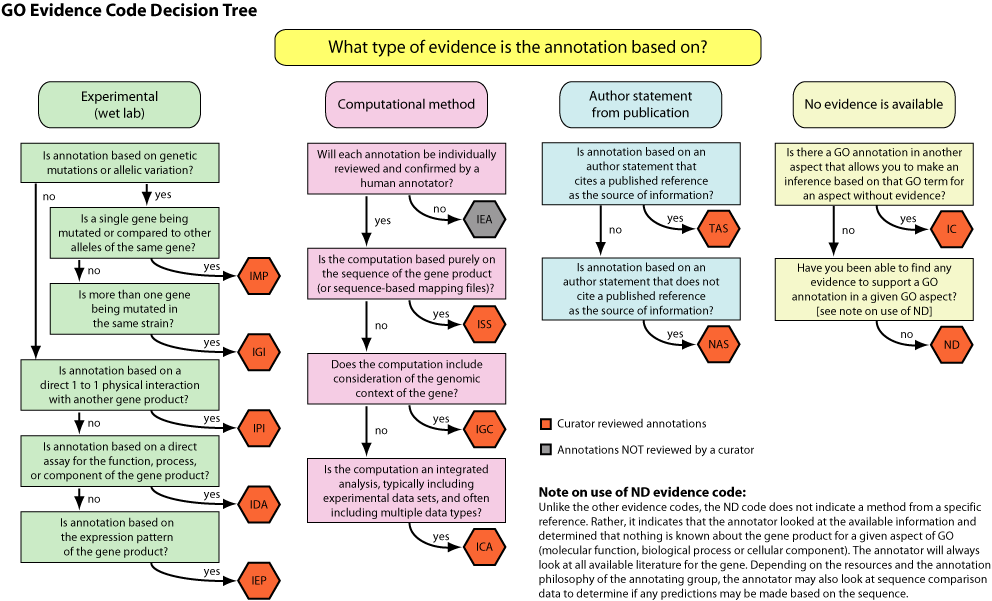
c. Les niveaux de preuve d'une annotation Ils sont précisés par des codes ("Evidence Codes") répartis en catégories : "Experimental" - "Computational" - "Author statement" (déclaration d'auteur) - "Curator statement" (déclaration de curateur)
Source : GO La dernière catégorie est "Automatically-assigned" (annotation automatique) dont le sous-code "Inferred from Electronic Annotation" (IEA) représente actuellement près de 95% de l'annotation. Voir : Skunca et al. (2012)
On ne soulignera jamais assez le le rôle primordial des scientifiques que l'on nomme curateurs. Ils effectuent, grâce à leur immense culture, un travail dans l'ombre qui assure la qualité, la rigueur et la pertinence des informations associées aux données de génomique, transcriptomique, protéomique et autres contenues dans les bases de données. GO est extrêmement complexe et nécessite un "navigateur" dans l'arbre de l'ontologie. Le plus utilisé pour GO est AmiGO. Voir un exemple de procédure d'annotation basée sur la séquence. |
|
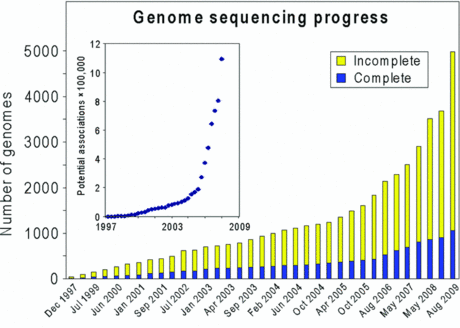
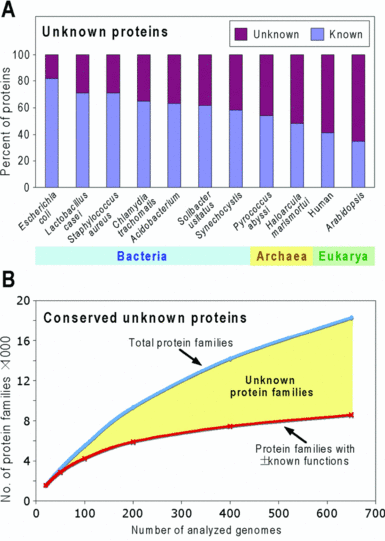
4. Les problémes de l'annotation a. Le décalage entre le nombre de génomes séquencés et leur documentation encyclopédique Le séquençage à trés haut débit génère des quantités phénoménales d'information. On peut donc considérer que l'annotation est maintenant le point d'achoppement des disciplines en "omique". En regard de la performance extraordinaire des technologies de ces domaines (séquençage d'un génome par jour) et donc l'accumulation tout à fait colossale de données de tous types dans les banques de données, on ne peut qu'induire un décalage entre les données brutes et leur interprétation, donc leurs significations biologiques. Tant que ce décalage existera, le pas suivant qui est d'extraire une "sur-information biologique" de cette information brute, ne pourra être correctement franchi.
Source : Hanson et al. (2010) |
| Exemple de l'évolution du nombre de génes estimés dans le génome humain | |||
| Technique | Date | Nombre de gènes estimés | Hypothèses et commentaires |
| "Calcul" initial | 1990 | 100 000 | Avec l'hypothèse que la taille moyenne d'un gène = 30 kb |
| Ebauche de séquençage du génome | 1994 | 71 000 | Résultat biaisé par les régions riches en gènes ? |
| Ilôts CpG | 80 000 | Avec l'hypothèse que 66% des gènes humains ont de tels "ilôts" | |
| Analyse des EST | 1994 | 64 000 | Gènes ayant un homologue dans GenBank - Redondance des EST de 50% |
| Chromosome 22 | 1999 | 45 000 | Correction liée à la haute densité en gène de ce chromosome |
| Technique "Exofish" ("Exon Finding by Sequences Homology") | 2000 | 28 000 - 34 000 | Avec l'hypothèse que les régions codantes sont plus conservées que les régions non-codantes. Comparaison des génomes homme - poisson ("Tetraodon nigroviridis") |
| EST | 2000 | 35 000 |
Nombre de gènes |
| Premier "brouillon" du génome | 2001 | 30 000 - 40 000 | Gènes connus + prédictions |
| Comparaison avec le génome de la souris | 2002 | 30 000 | Gènes connus + prédictions |
| Analyse du génome de l'homme en cours d'aboutissement | 2004 | 20 000 - 25 000 | Gènes connus + prédictions |
| 2007 | 20 000 | Annotation des gènes améliorée | |
| Analyse du génome de l'homme aboutie | 2012 | 20 687 gènes codant des protéines | "The ENCODE Project Consortium" |
| Source : Duret L. (2011) - "Bioinformatique: Annotation des génomes (eucaryotes)" | |||
|
b. Les erreurs ou l'absence d'annotation Les "inconnues" dans les bases de données :
Source : Hanson et al. (2010) Il est dommage d'accumuler une quantité inouie de données si on ne peut pas en tirer toute l'information. Ce déluge de données peut même "noyer" l'information 'actuelle" pertinente et nuire (au moins dans un premier temps). Quel intérêt d'obtenir à la suite d'une étude longue et coûteuse via des EST ou des puces à ADN, des informations telles que : "not annotated", "hypothetical protein" (!), "unnamed molecule, "putative function", ... On aboutit aux mêmes conclusions : X gènes sont sur-exprimés et Y sont sous-exprimés. Mais qui sont-ils, que font-ils, où sont adressées les protéines pour lesquelles ils codent ... ?! |
5. Les moyens pour l'annotation a. Exemples de logiciels et d'interfaces web pour l'annotation 1. Le consortium GO propose un ensemble de logiciels ("Gene Ontology Tools") pour traiter et analyser des données de divers types, en particulier celles issues des puces à ADN. Ces logiciels sont utilisables directement via une interface Web ou à installer sur l'ordinateur pour divers types de systèmes d'exploitation (Unix, Linux, Windows, Mac) 2. L'une des interfaces les plus didactiques et intuitives pour l'annotation : "QuickGO (GO Browser)" et "Gene Ontology Annotation (GOA)" (en particulier le lien : "Search GOA"). 3. Autres exemples de logiciels - interface web :
4. DAVID : outil pour annoter les données de transcriptome. 5. TD en ligne "Ontologies en biologie" - M2 Génomique Fonctionnelle - Université Aix - Marseille II |
b. Exemple dans le cas d'un gène Une fois un gène identifié, il faut obtenir le plus d'informations concernant :
Des logiciels bioinformatiques sont dédiées à l'étude de la structure des gènes et à leur annotation. Par exemple :
Des bases de données regroupent les données de structure des gènes et leur annotation :
Bien sur, l'ensemble de ces données sont intégrées dans les grandes bases de données biologiques mondiales que sont :
|
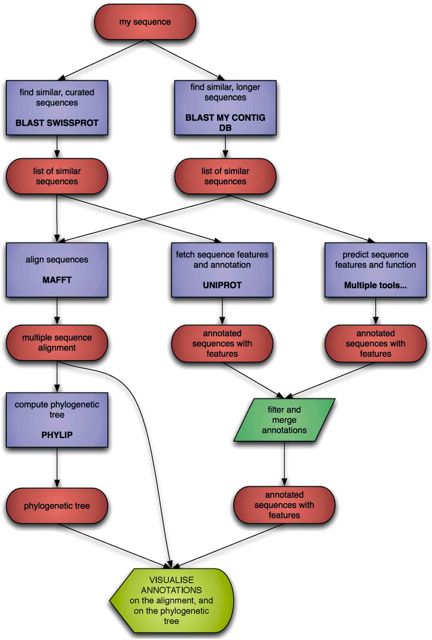
c. Les langages informatiques / Web L'annotation va de paire avec l'utilisation de langages spécifiques pour standardiser le format des données afin qu'ils soient transmis sans difficulté d'un service (logiciel) bioinformatique à un autre. De plus en plus, le langage XML avec des schémas de format XSD s'imposent. Figure ci-dessous : démarche logique du flux ("workflow") de données (ovales rouges) via un ensemble de services (logiciels) bioinformatiques (rectangles bleus). La transformation d'un type de données en un autre est d'autant plus performante que les formats en entrée et en sortie sont standardisés afin que n'importe quel logiciel puissent "accepter" les données entrantes (format d'échange commun BioXSD).
Source : Pettifer et al. (2010) d. Les autres moyens pour l'annotation L'ensemble de ces moyens fait partie de la génomique comparative et s'appuie sur la notion d'association. L'exemple typique est celui de génes bactériens regroupés en opéron codant les différentes enzymes qui catalysent les étapes d'une voie métabolique : la fonction d'un géne inconnu peut-être inferrée à partir des génes connus de cet opéron. Cette notion est étendue à la comparaison de génomes entiers. Une étude récente a ainsi permis de prédire la fonction de 19 familles de protéines d'Arabidopsis et de procaryotes (Gerdes et al., 2011). Parmi ces moyens, on peut citer :
D'autres sources d'informations (textuelles, images 2D, ...) peuvent être utilisées et combinées, soit manuellement, soit automatiquement. C'est le cas, par exemple, des méthodes d'annotation des gènes de l'embryon de souris (méthodes et base de données "EMAGE"). |
|
6. Quelques méthodes et outils pour l'annotation a. Généralités Elles sont de diverses natures, mais pour la plupart extrêmement théoriques. On conçoit intuitivement qu'il faut obtenir des outils logiciels qui puissent :
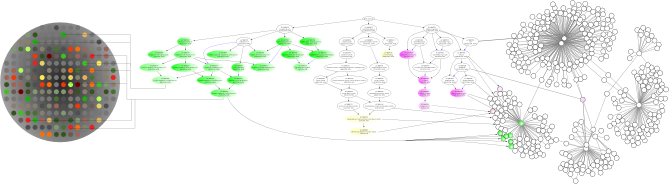
Source : "Ontologizer" Les avancées en bioinformatique (théoriques et/ou logicielles) permettent de plus en plus d'atteindre ce but. Dans l'exemple décrit ci-dessous, les auteurs décrivent une méthode de recherche de "catégories biologiques" contenant le plus d'informations liées aux gènes mis en évidence par telle ou telle approche de génomique et/ou de transcriptomique. Remarque : les auteurs emploient l'expression "biological categories". Que signifie-t-elle du point de vue de l'ontologie et encore plus de celui de la biologie ? Selon eux, le problème est que des bases de données telle que "Gene ontology" (GO) contiennent des milliers de "catégories" qui se chevauchent : l'obtention d'une "bonne catégorie" en renvoie un grand nombre d'autres qui y sont corrélées, ce qui n'aide pas à la prise de décision. Leur modèle (ci-dessous) analyse toutes les "catégories" d'une base de données d'ontologie en les insérant dans un réseau Bayesien ("model-based gene set analysis" - MGSA).
Source : Bauer et al. (2010) Au sein de ce réseau, la réponse de tel ou tel gène ("gene response" = sur-expression ou sous-expression ? Voir l'article) est modèlisée en fonction de "l'activation" de "catégories biologiques" identifiées par inférence probabilistique.
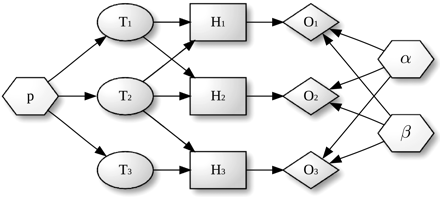
Leur résultat consiste à renvoyer la "meilleure catégorie" (augmentation de la précision) avec le moins de "catégories corrélées" (diminution du bruit de fond). Figure ci-dessous : l'algorithme qui traduit l'approche et le modèle décrits ci-dessus.
Source : Bauer et al. (2010) Voir l'application qui a été développée par ce groupe : "Graphviz". |
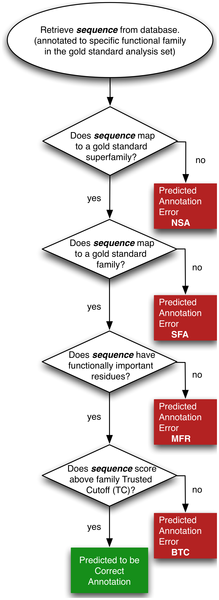
b. Des outils d'annotation via les motifs Les motifs sont des séquences consensus d'acides aminés qui signe une famille de protéines. Voir l'exemple du serveur Web "GOmotif" : entrer la séquence [GS]SSE.[DEG] (format "Prosite"). c. Des outils de ré-annotation D'autres outils bioinformatiques permettent de ré-annoter l'existant ou de confirmer l'annotation existante et ainsi de corriger les erreurs ou de donner des pistes pour une correction ultérieure. La figure ci-dessous présente le protocole d'analyse des erreurs d'annotation des fonctions des super familles d'enzymes dans les bases de données publiques.
Source : Schnoes et al. (2009) d. Des outils et des bases de données pour l'analyse des réseaux d'interactions proteines - protéines "PPI Finder" : un outil web pour la fouille de données textuelles des résumés publiés dans "PubMed" (NCBI) à la recherche de co-occurrences de mots et d'interactions entre ces mots, validées par leur existence dans les bases de données d'interactions protéines - protéines ("PPI databases") dont HPRD et BioGRID et les mots partagés de l'ontologie. e. D'autres outils en ligne
|
| f. Exemples de systèmes ou de bases de données d'annotation de génomes, de gènes, de voies métaboliques, de réseaux d'interactions etc ... | |
| "The gene ontology (GO) database" | GO current annotations |
| KAAS ("KEGG Automatic Annotation Server") | Moriya et al. (2007) "KAAS: an automatic genome annotation and pathway reconstruction server" Nucleic Acids Res. 35, W182-W185 |
| "The National Microbial Pathogen Data Resource's (NMPDR) Rapid Annotation using Subsystems Technology (RAST) server" | Aziz et al. (2008) "The RAST Server: Rapid Annotations using Subsystems Technology" BMC Genomics 9, 75 |
| "The Glimmer system" : suite logicielle pour le séquençage et l'assemblage de génomes, la recherche de gènes, l'annotation et l'analyse de génomes, l'analyse métagénomique (et autres outils génomiques et protéomiques) | "The Glimmer system" |
"Rice Genome Annotation Project" GeneDB : "genome database for prokaryotic and eukaryotic pathogens and closely related organisms" PomBase : "a comprehensive database for the fission yeast Schizosaccharomyces pombe, providing structural and functional annotation, literature curation and access to large-scale data sets" |
|
|
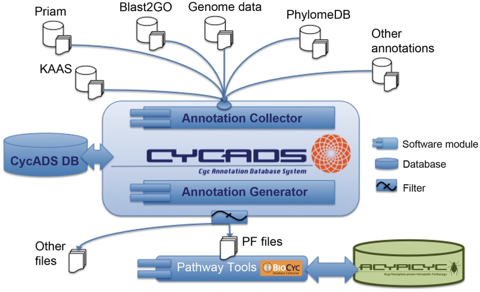
CycADS : "an annotation database system to ease the development and update of BioCyc databases" BioCyc : ensemble de bases de données (presque 3600 génomes et leurs voies métaboliques) |
Source : Vellozo et al. (2011) |
L'ontologie construite à partir du réseau d'interactions NeXO Les grands réseaux d'interactions entre gène et/ou entre protéines chez Saccharomyces cerevisiae permettent de déduire une ontologie dont la couverture et la puissance sont équivalentes à celles de Gene Ontology (GO), curée manuellement. L'ontologie extraites à l'aide de réseau ("Network-eXtracted Ontology" - NeXO) contient plus de 4000 termes biologiques et plus de 5700 relations entre ces termes, et inclue plus de 58 % des constituants cellulaires connus. Cet outil permet aussi de découvrir des relations entre termes non initialement cataloguées dans GO. Le profilage d'interactions par génétique quantitative et la chimiogénomique ont permis de caractériser des termes non identifiés par NeXO, notamment en relation avec des structures multimèriques liées à la fonction des protéines mitochondriales ou de trafic. L'approche NeXO permet de passer de l'utilisation d'ontologies pour évaluer des données à l'utilisation de données pour construire et évaluer des ontologies. |
Source : Dutkowski et al. (2013) |
| 7. Liens Internet et références bibliographiques | |
"Précis de Génomique" (2004) G. Gibson & S. Muse - Ed. De Boeck - ISBN : 2-8041-4334-1 Bettembourg, C. (2010) "Comparaison d'annotations GO entre plusieurs espèces" - Master 2 Modélisation des Systèmes Biologiques - Université de Rennes |
|
|
Bakke et al. (2009) "Evaluation of Three Automated Genome Annotations for Halorhabdus utahensis" PLoS ONE 4, e6291 Schnoes et al. (2009) "Annotation error in public databases: misannotation of molecular function in enzyme superfamilies" PLoS Comput. Biol. 5, e1000605 Pettifer et al. (2010) "The EMBRACE web service collection" Nuc. Acids Res. 38, W683 - W688 Hanson et al. (2010) "‘Unknown’ proteins and ‘orphan’ enzymes: the missing half of the engineering parts list – and how to find it" Biochem. J 425, 1-11 Gerdes et al. (2011) "Synergistic use of plant-prokaryote comparative genomics for functional annotations" BMC Genomics 12, S2 |
|
|
"The gene ontology (GO) database" Ashburner et al. (2000) "Gene Ontology: tool for the unification of biology" Nat. Genet. 25, 25 - 29 The gene ontology consortium (2001 ) "Creating the Gene Ontology Resource: Design and Implementation" Genome Res. 11, 1425 - 1433 Leonelli et al. (2011) "How the gene ontology evolves" BMC Bioinformatics 12, 325 |
|
|
"BioCyc" : ensemble de bases de données. Plus de 1100 génomes et leurs voies métaboliques. Vellozo et al. (2011) "CycADS: an annotation database system to ease the development and update of BioCyc databases" Database "CycADS : an annotation database system to ease the development and update of BioCyc databases" |
|
|
He et al. (2009) "PPI Finder: A Mining Tool for Human Protein-Protein Interactions" PLoS ONE 4, e455 Bauer et al. (2010) "GOing Bayesian: model-based gene set analysis of genome-scale data" Nuc. Acids Res. 1 -10 Skunca et al. (2012) "Quality of Computationally Inferred Gene Ontology Annotations" PLoS Comput. Biol. 8, e1002533 Logan-Klumpler et al. (2012) "GeneDB - an annotation database for pathogens" Nucleic Acids Res. 40, D98 - D108 Dutkowski et al. (2013) "A gene ontology inferred from molecular networks" Nature Biotech. 31, 38 - 45 |
|
|
"HPRD : Human Protein Reference Database" "BioGRID interaction database" "BioCreAtIvE : Critical Assessment of Information Extraction systems in Biology" |
|
![]()