
| Epigénétique - modification des nucléosomes et régulation de la transcription |
| Tweet |
|
|
1. Introduction
2. Les histones et leur association dans les nucléosomes 3. Modifications post-traductionnelles des histones 4. Signatures de la chromatine au niveau des gènes transcrits 5. Les protéines "writers"
6. Les protéines "erasers"
|
7. Domaines protéiques d'interaction avec les histones modifiées 8. La méthylation - déméthylation de l'ADN
9. Régulation par les ARN non codants
10 Méthodes pour étudier les modifications épigénétiques 11. Quelques abréviations et noms de protéines impliquées dans l'épigénétique 12. L'épitranscriptome : N6-méthyl-adénosine et autres nucléosides modifiés 13. Liens Internet et références bibliographiques |
|
1. Introduction L'ADN du génome haploïde de l'homme contient environ 3 milliards de paires de base. Chaque cellule diploïde (23 paires de chromosomes) de l'homme contient donc environ 6 milliards de paires de base.
En conséquence, l'ensemble des cellules du corps humain contient une longueur d'ADN qui correspond à 670 fois la distance soleil - terre ou 2,5 millions de fois le tour de la terre. Dans le noyau, l'ADN est donc extrêmement compacté, condensé et les gènes sont empaquetés dans la chromatine.
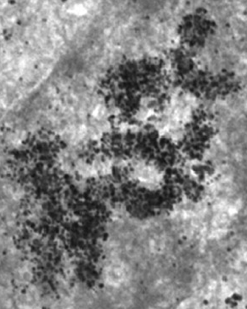
Source : Kireev et al. (2008) Les processus dynamiques de remodelage (condensation - décondensation / changement de structure) de la chromatine sont nécessaires pour l'étape initiale de la transcription des gènes. En effet, ce remodelage module l'accessibilité des protéines qui régulent et contrôlent la transcription aux promoteurs et aux régions régulatrices de la transcription ("enhancer", "silencer", "insulator", ...). a. Condensation de la chromatine - euchromatine et hétérochromatine L'ADN chromosomique est empaqueté à l'intérieur du noyau à l'aide des nucléosomes qui sont des complexes [ADN chargé négativement - protéines histone chargées positivement].
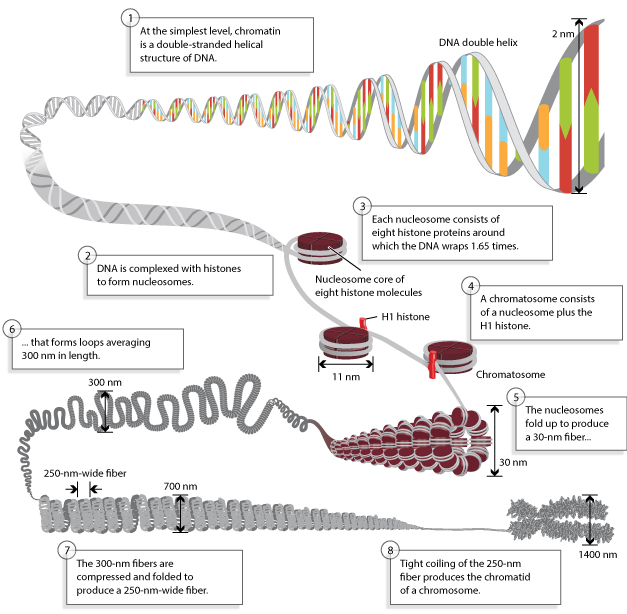
Source : Annunziato, Nature (2008) Un nucléosome est composé de huit protéines histone autour desquelles l'ADN (environ 146 - 147 paires de base) s'enroule 1,66 fois. C'est l'euchromatine (structure en collier de perles ou "beads on a string"). Les nucléosomes se replient pour former une fibre d'hétérochromatine de 30 nanomètres qui forme elle-même des boucles de longueur moyenne 300 nanomètres. Les boucles sont à leur tour compressées et repliées pour former une fibre de 250 nm d'épaisseur. Celle-ci est étroitement enroulée pour former les chromatides d'un chromosome. Les différences entre euchromatine et hétérochromatine sont beaucoup plus importantes que celà, tant du point de vue structural que fonctionnel. En particulier :
Source : Gaspar-Maia et al. (2011)
Autres facteurs qui ont un rôle sur l'état de condensation de la chromatine α. Les polyamines, en particulier la putrescine, la spermidine (4+) et la spermine (8+), sont des petits polycations aliphatiques linéaires impliquées dans de nombreuses fonctions biologiques. Les polyamines sont très présentes dans le noyau et elles ont des propriétés de compactage de la chromatine. En effet, ils semblent que les polyamines s'auto-assemblent avec des ions phosphates dans le noyau et génèrent des composés appelés agrégats de polyamines nucléaires ("Nuclear Aggregates of Polyamines") qui interagissent avec l'ADN génomique. La putrescine est synthétisée par l'ornithine décarboxylase et les deux autres polyamines sont des dérivés de la putrescine. β. Les protéines de maintien des chromosomes ("Structural Maintenance of Chromosomes (SMC) proteins") : leurs rôles n'est pas encore clairement défini mais elles ont une incidence sur la régulation de l'état de compacité de la chromatine au cours de certaines phases du cycle cellulaire (en particulier l'interphase). Les Eukaryotes possèdent au moins six protéines SMC différentes (Smc1 à Smc6) qui s'hétérodimèrisent sélectivement pour former trois complexes SMC :
La condensine contribue notablement à l'enroulement et au repliement progressifs des fibres de chromatine au moment de la préparation de la mitose. Elle participe également à la régulation de la transcription des gènes à l'interphase. L'état de condensation des fibres de chromatine résulte de deux activités : le super-enroulement dépendant de la condensine (condensation de la chromatine) et la relaxation induite par la topoisomérase (décondensation de la chromatine). |
b. Epigénétique et profils épigénétiques Une définition de l'épigénétique (épi = autour, en plus) : régulation de la transcription sans modification de la séquence de l'ADN. De plus en plus de définitions du mot "épigénétique" sont proposées avec les avancées dans ce domaine (en particulier liées à l'héritabilité de phénotypes). Les facteurs épigénétiques responsables du processus de remodelage de la chromatine sont :
Contrairement à la séquence de l'ADN, qui est en grande partie inchangée au cours de la vie, les profils épigénétiques varient non seulement d'un tissu à un autre mais changent avec l'âge et sont sensibles aux influences comportementales et environnementales (protection maternelle, pollution, qualité de la nutrition, diète, alcool, cigarette, stress, ...). Les profils de méthylation de l'ADN en particulier (et les modifications épigénétiques en général) semblent spécifiques :
Les mécanismes épigénétiques contribuent donc à l'identité de chaque type de cellule (plus de 200 types de cellule ont été recensés chez l'homme). Les protéines très conservées du groupe Polycomb (PcG) et du groupe Trithorax (TrxG) peuvent conférer une mémoire héritable à long terme : les deux phases du cycle cellulaire au cours de laquelle la "mémoire épigénétique" est la plus susceptible d'être effacée sont la réplication de l'ADN et la mitose.
Ces changements de concentration des protéines et de volume de la chromatine ont des effets importants sur la cinétique de fixation des protéines qui contrôlent la structure de la chromatine. Les protéines PcG et TrxG se fixent sur la chromatine au cours de la mitose mais les protéines TrxG restent davantage fixées. L'acétylation et l'ubiquitinylation des histones est réduite au cours de la mitose, les méthylations H3K4 et H3K27 sont inchangées et certaines phosphorylations sont accrues. À l'échelle qui s'échelonne des kilo- paires de bases aux méga- paires de bases (échelle qui englobe la taille des gènes, des groupes de gènes et des domaines de régulation), l'organisation tridimensionnelle de l'ADN est impliquée dans les mécanismes de régulation de la transcription des gènes. A cette échelle, le génome est subdivisé en domaines qui sont dans différents états épigénétiques : états actifs du point de vue de la transcription, états inactifs ou états réprimées par des protéines du groupe Polycomb (Boettiger et al., 2016). Début 2015, le "NIH Roadmap Epigenomics Consortium" a généré et analysé la plus grande collection d'epigénomes humains de cellules primaires et de tissus (analyse intégrative de 111 épigénomes humains de référence). Ces données ont apporté un très grand nombre d'informations quant aux profils de modification des histones, à l'accessibilité et la méthylation de l'ADN, à l'expression des ARN. Ces données ont permis d'établir des cartes globales d'éléments régulateurs et de définir des modules régulateurs de l'activité coordonnée avec leurs activateurs et leurs répresseurs probables. L'ensemble des résultats démontrent que des variants génétiques associés à des traits et à des maladies sont enrichis en marques épigénomiques spécifiques des tissus, ce qui établit une corrélation entre des types de cellules biologiquement pertinents et divers traits humains. Roadmap Epigenomics Consortium et al. (2015) "Integrative analysis of 111 reference human epigenomes" Nature 518, 317–330 |
| 2. Les histones et leur association dans les nucléosomes |
|
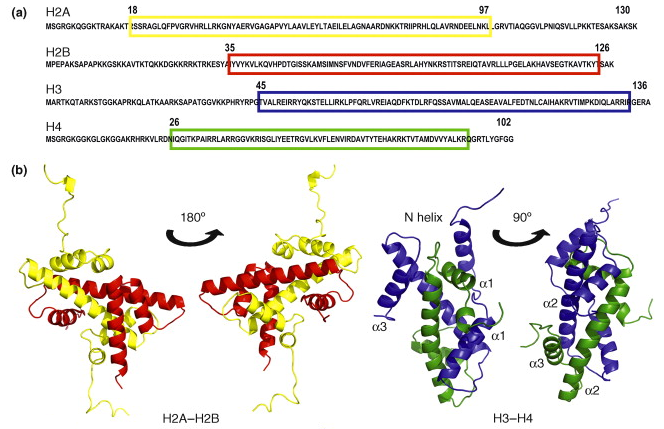
Il existe 5 familles d'histones : H1/H5, H2A, H2B, H3 et H4. Chaque famille contient plusieurs sous-familles qui contiennent plusieurs types d'histones. Les histones (entre 13 et 15 kDa) contiennent beaucoup d'acides aminés basiques. |
||||||
| Famille | Histones H1 | Histones H2A | Histones H2B | Histones H3 | Histones H4 | Histones H5 |
| Acides aminés (dépend des variants) | 194 - 255 | 130 | 126 | 135 - 136 | 102 - 103 | 190 |
| Masse molaire (kDa) | 21 - 29 | 14 | 14 | 15 - 16 | 11,3 | 21 |
| Sous-familles (la nomenclature peut varier d'une source à l'autre) | H1F - H1H1 | H2AF - H2A1 - H2A2 | H2BF - H2B1 - H2B2 | H3A1 - H3A2 - H3A3 | H41 - H44 | ------ |
| Nombre de membres dans la famille (octobre 2014) | 126 | 265 | 214 | 216 | 116 | ------ |
| Nombre de gènes codant chez l'homme | 10 | 26 | 23 | 18 | 14 | ------ |
| Lien Uniprot vers un membre d'une sous-famille | Q02539 | P0C5Y9 | Q96A08 | P68431 | P62805 | P02259 |
|
Le variant macro-H2A contient 327 acides aminés (40 kDa) et il a des fonctions spécifiques dans l'inactivation du chromosome X et la régulation de la transcription. Ontologie : GO:0006334 - Biological process : nucleosome assembly |
||||||
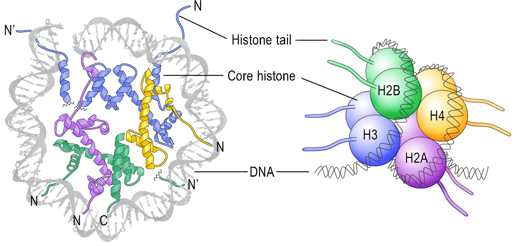
Les histones H2A, H2B, H3 et H4 ("core histones") forment le coeur de la structure octamérique impliquée dans les nucléosomes. Les histones H1 et H5 sont appelées histones de liaison ("linker histones").
Source : Graff & Mansuy (2008) Le nucléosome contient 2 molécules de chaque histone H2A, H2B, H3 et H4 assemblées en deux hétérodimères [H2A-H2B] et un hétérotétramère [H3-H4]. Les histones adoptent un repliement qui leur est caractéristique ("histone fold") : 3 hélice α reliées par 2 boucles. Les histones forment des dimères via des interactions entre leurs longues hélices α2 (orientation anti-parallèle)
Source : Das et al. (2010) Deux dimères [H3-H4] forment un faisceau de 4 hélices stabilisé par des interactions [H3-H3’]. L'hétérodimère [H2A/H2B] se fixe à l'hétérotétramère [H3-H4] via des interactions entre H2B et H4. Interactions inter-nucléosomes dans les fibres de chromatine isolées Les nucléosomes adjacents sont liés par une partie de l'ADN appelé ADN de liaison ("linker DNA") dont la longueur varie de 10 à 80 paires de base selon l'espèce ou le type de tissu. La longueur moyenne de l'ADN de liaison chez les vertébrés est 35 paires de bases. Les études (Shogren-Knaak et al., 2006) des effets de la perte de la charge positive de K16 de l'histone H4 (par acétylation) ont montré l'importance des contacts inter-nucléosomes entre l'extrémité N-terminale (chargée positivement) de l'histone H4 et la partie riche en acides aminés acides d'une partie de l'histone H2A. Rôle physique de chaque extrémité N-terminale d'histones dans le repliement de la chromatine (Arya & Schlick, 2006)
Ces interactions induisent collectivement un arrangement global hélicoïdal en "zig-zag" où les nucléosomes interagissent fortement avec leurs quatrième nucléosome voisin. Rôle des histones au sein des nucléosomes
Les histones de liaison sont associées à l'ADN de liaison. Elle protègent partiellement (environ 20 paires de bases) l'ADN de liaison de l'action des nucléases. L'histone H1 contient une région globulaire conservée et une extrémité C-terminale riche en lysines capable d'interagir fortement avec l'ADN. |
|
Visualisation du nucléosome de Xenopus laevis à une résolution de 2,8 Å Code PDB : 1AOI Complexe histones [H3,H4,H2A,H2B] et un fragment d'ADN de 146 paires de base.
|
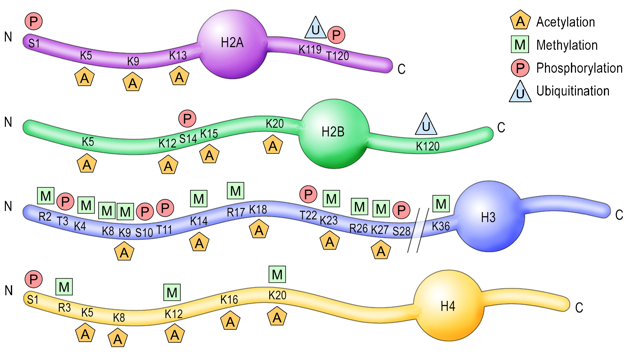
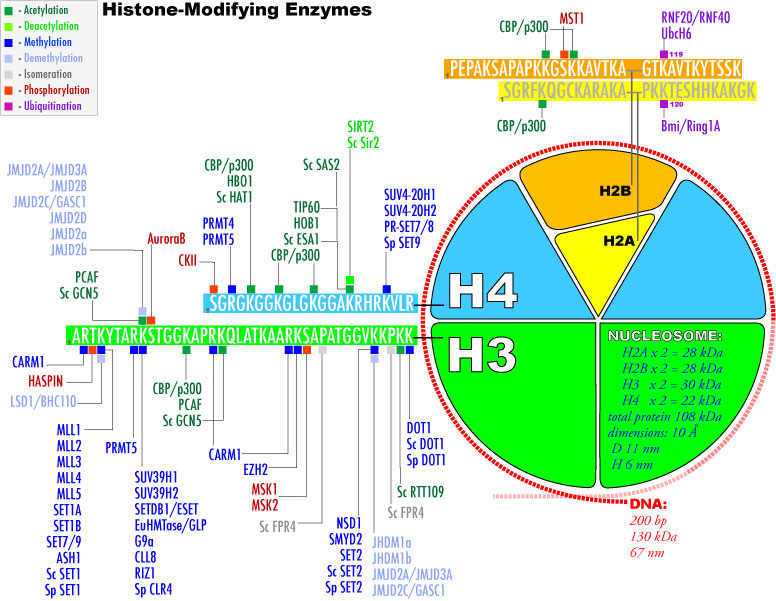
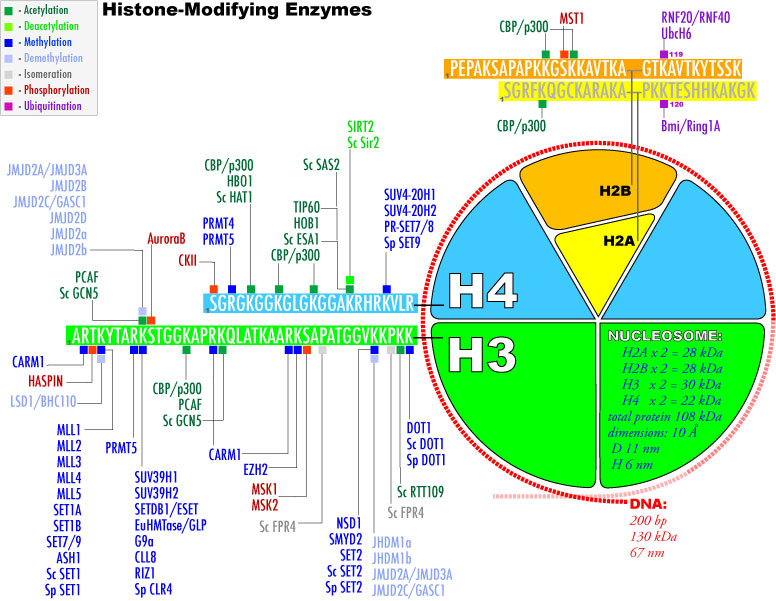
3. Modifications post-traductionnelles des histones L'extrémité N-terminale de la chaîne polypeptidique des histones qui émerge des nucléosomes subit de nombreuses modifications post-traductionnelles.
Source : Graff & Mansuy (2008) |
| Acide aminé | groupe ε-aminé de lysines | groupe guanidinyle d'arginines | sérines, thréonines et tyrosines | histidines (plus rare) |
| Modifications post-traductionnelles | mono-, di- et triméthylation , acétylation, ubiquitinylation, sumoylation, crotonylation, poly(ADP)-ribosylation |
mono-méthylation ou di-méthylation symétrique ou asymétrique |
phosphorylation |
mono-méthylation |
| Remarque | Les sites de méthylation des lysines des histones les plus étudiés sont H3 lysine 4 (H3K4), H3K9, H3K27, H3K36, H3K79 et H4K20. | Les sites de méthylation des arginines des histones les plus étudiés sont H3R2, H3R8, H3R17, H3R26 et H4R3. | ------------ | |
| Cependant, les études de spectromètrie de masse et de protéomique quantitative ont montré que d'autres acides aminés basiques des histones H1, H2A, H2B, H3 et H4 sont méthylés. | ||||
| Autres modifications observées |
|
|||
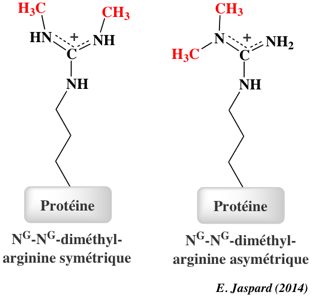
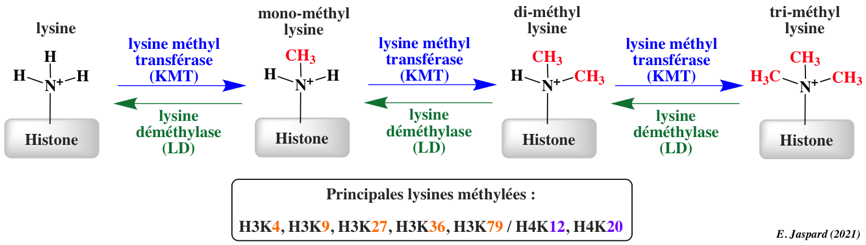
La méthylation des histones se produit principalement sur les chaînes latérales des lysines et arginines. Contrairement à l'acétylation et la phosphorylation, la méthylation ne modifie pas la charge des histones. Il y a un niveau supplémentaire de complexité en ce qui concerne la méthylation :
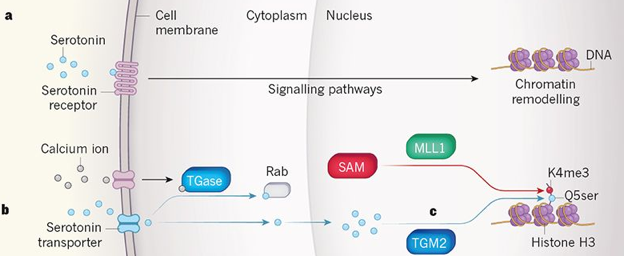
La phosphorylation et l'acétylation des histones diminuent la charge positive des histones ce qui diminue les interactions électrostatiques entre l'ADN et les histones. La chromatine est moins compacte et l'accessibilité des protéines de la machinerie de la transcription est augmentée. L'ubiquitinylation ajoute une très grosse molécule aux histones, induisant ainsi un profond changement de la conformation général du nucléosome, qui à son tour modifie les interactions intra-nucléosomique et/ou les interactions avec d'autres complexes liés à la chromatine. La coupure de l'extrémité N-terminale saillante des histones (perte des 21 premiers acides aminés de H3) a des effets similaires. La sérotonylation
Source : Cervantes & Sassone-Corsi (2019) c. Dans le noyau :
Les modifications post-traductionnelles des histones ont des incidences les unes sur les autres (elles peuvent s'exclure ou au contraire s'induirent mutuellement). Cette interférence entre les différentes modifications post-traductionnelles contribue probablement à affiner le contrôle étroit de la structure de la chromatine. |
4. Signatures de la chromatine au niveau des gènes transcrits La tri-méthylation de la lysine 4 de l'histone H3 (H3K4me3) est une modification caractéristique de la chromatine au niveau des promoteurs qui contrôlent des gènes activement transcrits chez les eucaryotes (de la levure à l'homme). Cette modification s'effectue sur les nucléosomes bordant des régions sans nucléosome et qui coïncident avec les sites de démarrage de la transcription ("transcription start sites" - TSS) des gènes transcrits. Les modifications H3K4me1 (mono-méthylation), H3K4me3 (tri-méthylation) et H3K36me3 (tri-méthylation) sont les marques respectives des :
Le complexe "Set1-containing COMPASS" est recruté sur les promoteurs "actifs" via le facteur d'élongation Paf1 ("RNA polymerase II-associated factor 1"). Ce complexe occupent les régions activement transcrites et son recrutement dépend aussi de la forme active de l'ARN polymérase II (dont la sérine 5 est phosphorylée). L'histone méthyltransférase SET1 méthyle spécifiquement la lysine 4 de l'histone H3 sauf si la lysine 9 est déjà méthylée. Elle catalyse la réaction : S-adénosyl-L-méthionine + L-lysine-[histone] ===> S-adénosyl-L-homocysteine + N(6)-méthyl-L-lysine-[histone]. La tri-méthylation H3K4me3 au niveau des promoteurs "actifs" nécessitent une autre modification d'histone : la mono-ubiquitinylation de la lys 123 de l'histone H2B catalysée par [Rad6- Bre1]. Deux familles de protéines (le groupe trithorax et le groupe Polycomb) contiennent des protéines associées à la chromatine et qui participent à l'activation ou à la répression de la transcription. Les protéines de ces deux familles contiennent un motif de 130 - 140 acides aminés, appelé domaine SET ["Su(var)3-9, Enhancer of zeste [E(z)] and trithorax (trx)"]. Voir : "The Polycomb and Trithorax page". COMPASS ("Complex Proteins Associated with Set1"), est un complexe de 7 chaînes polypeptidiques (de 25 à 130 kDa). Il existe au moins six homologues de COMPASS chez les mammifères (les complexes MLL1-4, hSET1A et hSET1B). |
| Exemples de régulation de la transcription via la modification d'histone | ||||||||
| modification | H2BK5 | H3K4 | H3K9 | H3K14 | H3K27 | H3K79 | H3K36 | H4K20 |
| mono-méthylation | activation | activation | activation | ----- | activation | activation | ----- | activation |
| di-méthylation | ----- | ----- | répression | ----- | répression | activation | ----- | ----- |
| tri-méthylation | répression | activation | répression | ----- | répression | activation répression |
activation | ----- |
| acétylation | ----- | ----- | activation | activation | activation | ----- | ----- | ----- |
L'acétylation H3K27 régule la cinétique de la transcription :
|
||||||||
| Source : Wikipédia | ||||||||
La phosphorylation de Tyr 57 (hautement conservée) de l'histone H2A par par l'activité tyrosine kinase insoupçonnée de la caséine kinase 2 (CK2) régule l'étape d'allongement de la transcription. Ce processus est conservé de la levure aux mammifères. CK2α (la sous-unité catalytique de CK2) se fixe sur des gènes transcrits par l'ARN polymérase II et des "enhancers" actifs. Du point de vue du mécanisme, l'inhibition de CK2 ou la mutation Y57F de l'histone H2A augmente l'activité de dés-ubiquitinylation (de l'histone H2B) par le complexe acétyltransférase Spt-Ada-Gcn5 (SAGA). Ce résultat suggère un rôle essentiel de cette phosphorylation dans la coordination de l'activité du complexe SAGA lors de la transcription. Voir Basnet et al. (2014). |
5. Les protéines "writers" a. Les histone lysine acétyltransférases (HAT) Il existe deux grandes classes de HAT (EC 2.3.1.48) :
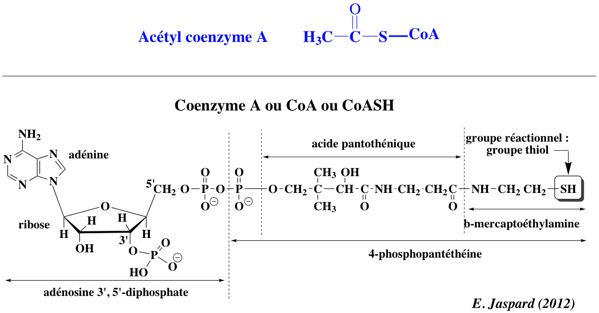
Voir un tableau des abréviations et noms de protéines impliquées dans l'épigénétique. Réaction catalysée : acétyl-CoA + [histone] <=> CoA + acétyl-[histone] Les HAT utilisent l'acétyl-CoA comme cofacteur et catalysent le transfert d'un groupe acétyle sur le groupement ε-aminé des chaînes latérales de lysine.
La charge positive de la lysine est neutralisée ce qui affaiblit les interactions entre les histones et l'ADN. b. Les histone lysine méthyltransférases (HKMT) Il existe deux types de HKMT (EC 2.1.1.43) :
Voir un tableau des abréviations et noms de protéines impliquées dans l'épigénétique.
Toutes les HKMT catalysent le transfert d'un groupement méthyle de la S-adénosylméthionine sur le groupement ε-aminé d'une lysine : S-adénosyl-L-Met + histone L-Lys => S-adénosyl-L-homocysteine + histone N6-méthyl-L-Lys
La dérégulation des lysine méthyltransférases est associée à de nombreuses maladies. Des progrès significatifs ont été réalisés dans le développement de médicaments qui ciblent ces enzymes.
c. Les histone arginine méthyltransférases (PRMT) Ces enzymes (EC 2.1.1.125) catalysent le transfert d'un groupement méthyle de la S-adénosylméthionine sur le groupe ω-guanidino de l'arginine : S-adénosyl-L-Met + Arg-[histone] <=> S-adénosyl-L-homocysteine + N(omega)-méthyl-Arg-[histone] Il existe deux classes de PRMT :
Exemples de spécifcité :
Voir un tableau des abréviations et noms de protéines impliquées dans l'épigénétique. d. Autres types de protéines "writers" : arginine déiminases, lysine biotinases, lysine ribosylases, lysine ubiquitinases, sérine/thréonine/tyrosine kinases. Voir la base de données :"HIstome". |
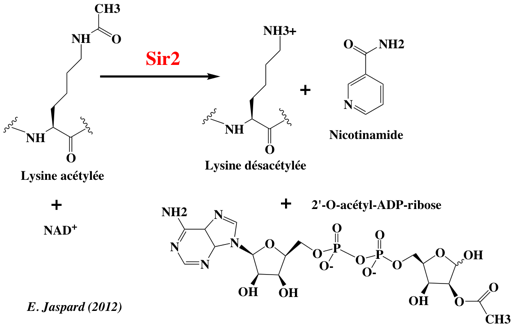
6. Les protéines "erasers" a. Les histone lysine désacétylases (HDAC) Les HDAC réversent l'acétylation de la lysine par les HAT, ce qui restaure la charge positive de la lysine. Il existe quatre classes d'HDAC (EC 3.5.1.98) :
Voir un tableau des abréviations et noms de protéines impliquées dans l'épigénétique. b. Les histone lysine déméthylases (HLD) Il existe deux classes principales d'HLD :
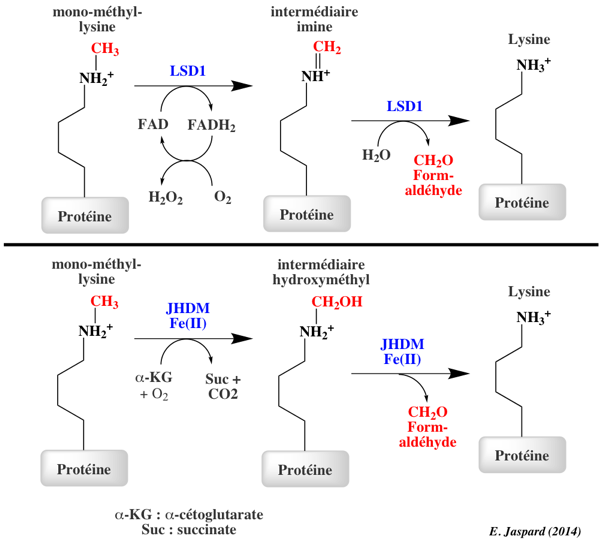
Les histone lysine déméthylases ont des spécificités précises. La première histone lysine déméthylase à FAD identifiée (en 2004) et a été nommée "Lysine-specific demethylase 1" (LSD1 aussi appelée KDM1A). Elle :
La "Lysine-specific demethylase 4A" ou JHDM3A :
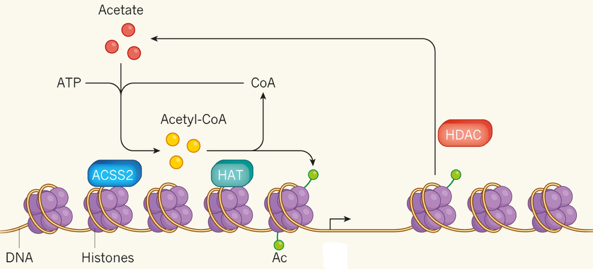
La "Lysine-specific demethylase 6A" - KDM6A déméthyle spécifiquement K27 di- et tri-méthylée de l'histone H3 mais ne déméthyle pas K27 mono-méthylée. La déméthylation de K27 est concomitante avec la méthylation K4 de l'histone H3 et elle régule le recrutement du complexe PRC1 et la mono-ubiquitinylation de l'histone H2A. Voir un tableau des abréviations et noms de protéines impliquées dans l'épigénétique. c. Les histone arginine déméthylases (HRDM) Les HRDM sont des dioxygénases nécessitant Fe(II) et l'α-cétoglutarate comme cofacteurs et qui peuvent agir en tant qu'histone arginine déméthylase ou en tant que lysyl-hydroxylase (voir histone lysine déméthylases). Exemple de spécificité : JMJD6 ("Jumonji domain-containing 6 protein") déméthyle R2 de l'histone H3 et R3 de l'histone H4. d. Autres types d'enzymes "erasers" : lysine déribosylase, lysine déubiquitinases, sérine/thréonine/tyrosine phosphatases. Voir la base de données :"HIstome". e. Synthèse d'acétyl-CoA et modulation de la transcription L'acétyl-coenzyme A (acétyl-CoA) est synthétisé par l'acétyl-CoA synthétase 2 (ACSS2) à partir d'acétate, de coenzyme A (CoA) et d'ATP.
Source : Ashley Watson & Tsai (2017) Chez la souris, ACSS2 se fixe sur la chromatine dans des neurones différenciés ou matures. ACSS2 se fixe à proximité de séquences d'ADN qui contrôlent la transcription de gènes qui régulent la différenciation ou la consolidation de la mémoire à long terme.
|
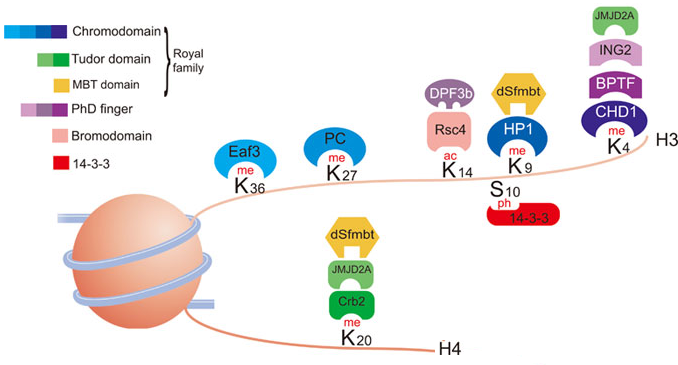
7. Domaines protéiques d'interaction avec les histones modifiées Il existe un très grand nombre de protéines associées à la chromatine. Elles interagissent spécifiquement avec les histones modifiées par l'intermédiaire de nombreux domaines distincts.
Source : Bannister & Kouzarides (2011) Ces domaines permettent la reconnaissance simultanée de plusieurs types de modifications post-traductionnelles et d'autres fonctions des nucléosomes. Les types de domaines reconnaissant la méthylation de la lysine sont les plus nombreux, reflétant probablement l'importance relative de cette modification post-traductionnelle. |
| Méthyl-lysine et lysine | Méthyl-arginine et arginine | Acétyl-lysine | Phosphosérine |
| Module "Royal family" : chromodomaine, domaine Tudor et domaine PWWP Plant homeodomain (PHD) finger Bromo-adjacent homology (BAH) domains ATRX-DNMT3A-DNMT3L (ADD) domain Tandem Tudor domains |
Canonical and expanded Tudor domains PHD Finger Domains WD40 Repeat Domains |
Bromodomains and Tandem Bromodomains TRIM Family Bromodomains Tandem PHD Fingers Tandem PH Domains Bromodomain and extraterminal (BET)-Family Bromodomains |
Protéines 14-3-3 Tandem BRCT Domains The baculovirus IAP repeat (BIR) domain of survivin |
| Voir un tableau des abréviations et noms de protéines impliquées dans l'épigénétique. | |||
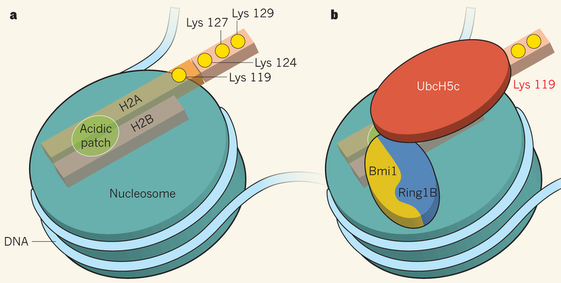
Exemple de mécanisme de reconnaissance entre enzyme de modification des histones et nucléosome PRC1 ("Polycomb repressive complex 1") agit avec les autres complexes protéiques du groupe Polycomb pour réprimer la transcription de nombreux gènes qui contrôlent les processus du développement chez les animaux. PRC1 peut compacter la chromatine et inhiber le remodelage d'un nucléosome par un mécanisme non enzymatique. Cependant, PRC1 semble agir également par un processus enzymatique d'ubiquitinylation. Six formes de PRC1 ont été identifiées chez les mammifères : toutes contiennent deux protéines qui possèdent un domaine structural appelé RING ("Really Interesting New Gene"). Ce type de domaine lie le zinc et est souvent trouvé dans la structure de protéines impliquées dans l'ubiquitinylation. Une structure cristallographique (McGinty et al., 2014) montre que l'association des enzymes [Ring1B - Bmi1] qui fait partie du complexe répressif PRC1, positionne une autre enzyme, UbcH5c, juste au-dessus de Lys 119 de l'histone H2A, permettant la mono-ubiquitinylation de Lys 119. Le schéma ci-dessous montre le nucléosome et la partie de caractère acide ("acidic patch") formée par les histones H2A et H2B.
Source : Müller & Müller (2014) Lys 119, Lys 124, Lys 127 et Lys 129 de l'histone H2A sont indiquées par des cercles jaunes. Ring1B : enzyme "Ubiquitin ligase E3"; Bmi1 : "Polycomb-group RING finger (PCGF) protein"; UbcH5c : enzyme "Ubiquitin-conjugating enzyme E2" La Lys 119 mono-ubiquitinylée crée alors un site de fixation pour le complexe histone-méthyltransférase PRC2 et favorise ainsi l'addition de 3 groupes méthyles sur Lys 27 de l'histone H3, cette modification étant capitale pour la répression de la transcription des gènes cibles de Polycomb. |
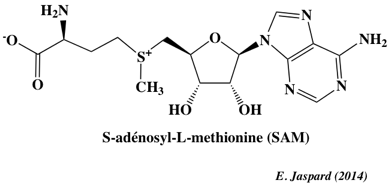
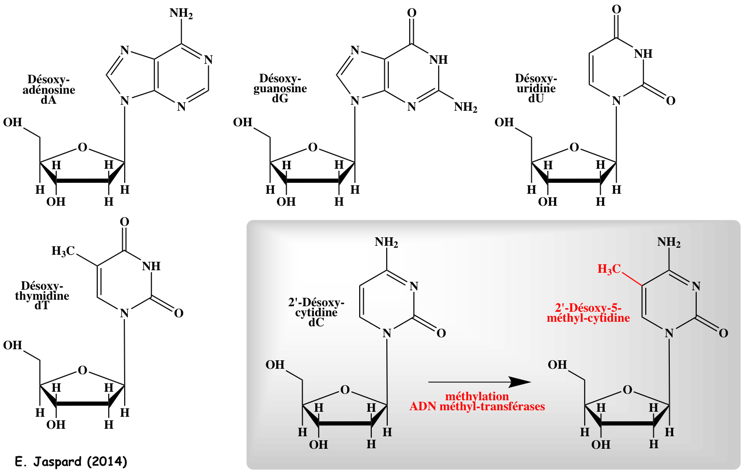
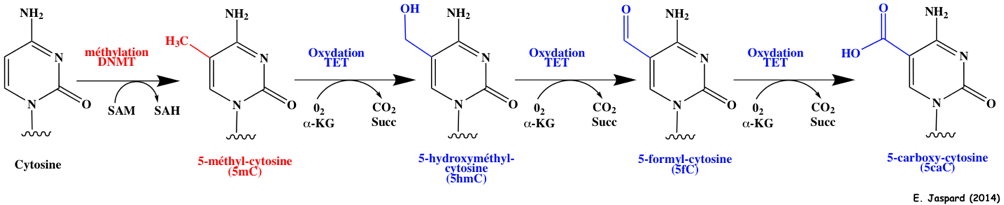
8. La méthylation - déméthylation de l'ADN a. Formation de la 5-méthyl-cytosine (5mC) L'autre grande modification épigénétique est la méthylation de l'ADN. Elle est essentielle pour la régulation de la transcription, pour éteindre les éléments rétroviraux, pour l'empreinte génomique et pour l'inactivation du chromosome X, ... La méthylation de l'ADN est catalysée par une famille d'ADN méthyltransférases ("DNA methyltransferases" - DNMT) qui transfèrent un groupe méthyle de la S-adénosyl-méthionine (SAM) sur le carbone en position 5 d'une cytosine pour former la 5-méthyl-cytosine (5mC).
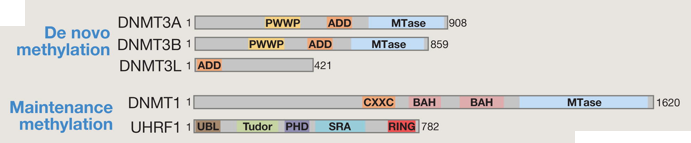
Méthylation d'entretien L'ADN méthyltransférase DNMT1 se positionne au niveau de la fourche de réplication de l'ADN et copie le profil de méthylation de l'ADN parental sur le brin néo-synthétisé. Elle est capable de réparer les erreurs de méthylation de l'ADN. Elle est aussi appelée méthylation d'entretien ("maintenance DNMT"). Méthylation de novo Les ADN méthyltransférases DNMT3a et DNMT3b (de structure similaire à DNMT1) peuvent mettre en place un nouveau profil de méthylation de l'ADN non modifié et sont ainsi appelées "de novo DNMT". Leur activité peut-être modulée par un membre de cette famille, catalytiquement inactif : DNMT3L. Domaines fonctionnels prédits au sein des protéines (de souris) impliquées dans la méthylation de novo et la méthylation d'entretien.
Source : Wu & Zhang (2014) L'ADN méthylé est reconnu par 3 familles de protéines qui sont les partenaires obligatoires des DNMT :
La déméthylation Domaines fonctionnels prédits au sein des protéines (de souris) impliquées dans l'oxydation de la 5mC et l'excision [5fC / 5caC].
Source : Wu & Zhang (2014)
Méthylation et déméthylation par oxydations successives de la 5mC par les protéines TET ("Ten-Eleven Translocation") :
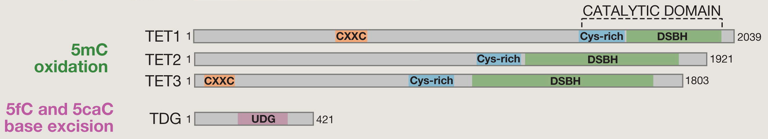
Toutes les protéines TET possèdent un domaine catalytique C-terminal qui contient une région riche en Cys et un grand domaine à hélice β double brin (DSBH - "Double-Stranded β-Helix"). Les protéines TET1 et TET3 contiennent également un domaine N-terminal en doigt de zinc (motif CXXC). L'excision des bases ("base-excision repair" - BER) est catalysée par l'ADN thymine glycosylase ("Thymine DNA glycosylase" - TDG) qui contient un domaine "Uracile DNA glycosylase" (UDG). Exemples d'autres modifications covalentes de l'ADN
|
|
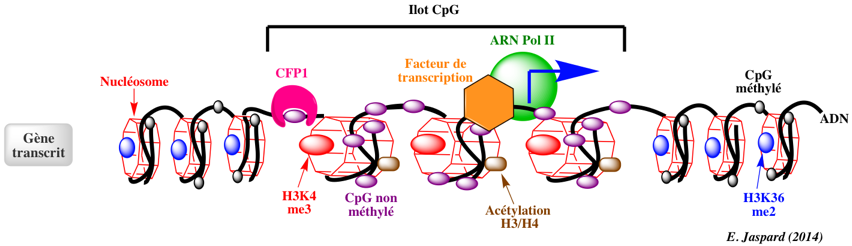
L'essentiel des sites de méthylation de l'ADN se situent sur des cytosines qui précèdent une guanine (CpG - le "p" signifie phosphate). Les ilots CpG sont des fragments d'ADN d'au moins 200 paires de base, riches en GC et dont la proportion en CpG est plus élevée que dans le reste du génome. Ils contiennent moins de nucléosome. Les génomes des mammifères sont généralement caractérisés par un grand déficit en CpG (sous-représentés). Par exemple, le rapport [CpGobservés / CpGattendus] est d'environ 0,20 - 0,25 dans les génomes de l'homme et de la souris. En effet :
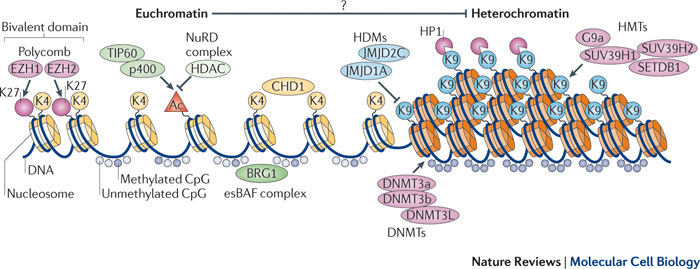
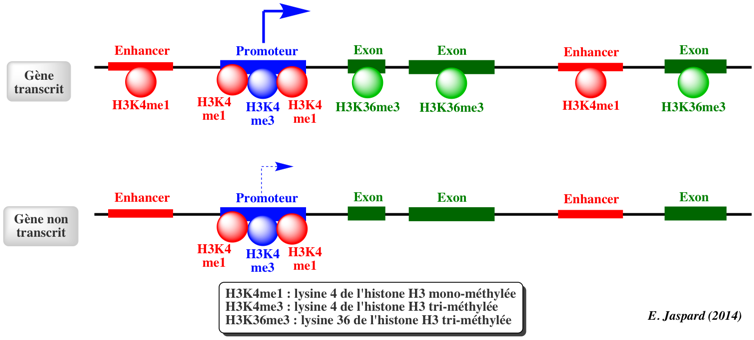
Dans le génome humain, 60% à 80% des 28 millions d'ilots CpG recensés sont méthylés dans les cellules somatiques. Chez les mammifères, une proportion importante des promoteurs se situent dans des ilots CpG et/ou possèdent une forte teneur en CpG. Certains sites non-CpG peuvent être aussi méthylées mais la méthylation des cytosines de type CpH (H = A, T ou C) a lieu surtout chez les plantes et très peu chez les mammifères. Le schéma ci-dessous illustre l'état de la chromatine au niveau des ilots CpG pour des gènes transcrits.
Les ilots CpG pour des gènes transcrits sont généralement :
Facteurs de transcription sensibles à la méthylation de leur site de fixation Certains motifs de fixation à l'ADN n'ont pas encore de facteurs de transcription identifiés : c'est le cas notamment de l'élément CGCG associé à des gènes fortement transcrits dans les tissus humains et très présent près du site de début de transcription d'un sous-ensemble d'ilots CpG. La protéine nucléaire associée à BTG3 ("BTG3-Associated Nuclear Protein" - BANP) est un facteur de transcription qui se fixe sur cet élément dans le génome de la souris et de l'homme :
|
9. Régulation par les ARN non codants a. La méthylation de novo de l'ADN dirigée par les siARN chez les plantes Les noyaux des Eucaryotes contiennent 3 complexes ARN polymérases ADN-dépendantes [nucléoside triphosphate + ARN(n) => diphosphate + ARN(n+1)]:
Les plantes possèdent 2 ARN polymérases supplémentaires appelées Pol IV et Pol V, toutes deux ayant évolué à partir de Pol II et qui sont spécialisés dans la méthylation de l'ADN dirigée par les ARN ("RNA-directed DNA methylation" - RdM). Pol II, Pol IV et Pol V sont constituées de 12 sous-unités dont beaucoup sont communes à ces trois polymérases. Chacune possède également des sous-unités qui leur sont spécifiques. Ces sous-unités sont nommés ARN polymérase nucléaire B ("Nuclear RNA Polymerase B " - NRPB) pour les sous-unités spécifiques de Pol II, NRPD pour les sous-unités spécifiques de Pol IV et NRPE pour les sous-unités spécifiques de Pol V. Schématiquement, les étapes de la RdM sont les suivantes :
Voir un cours sur l'interférence ARN (siARN et miARN). Ce processus d'une très grande complexité, dont on commence à décrypter les mécanismes, met en jeu des dizaines de protéines. Par exemple :
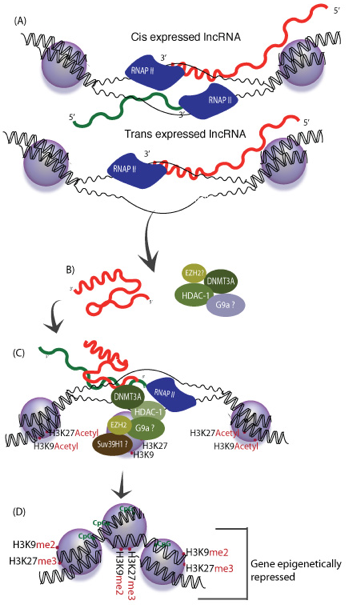
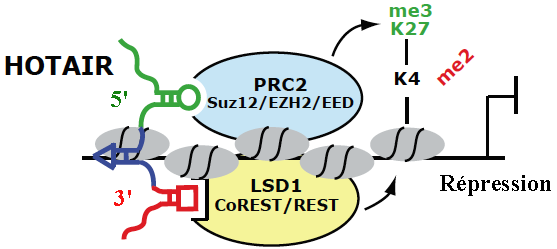
En absence de la protéine "Decreased DNA Methylation 1" (DDM1), une voie RdDM non-classique impliquant Pol II et RDR6 génère des ta-siRNAs ("21 nucleotide trans-acting small interfering RNA" - dérivés de transcrits endogènes TAS1, TAS2 et TAS3) qui guident la méthylation de novo de l'ADN. Voir une revue : Matzke & Mosher (2014) b. Régulation épigénétique par les longs ARN non-codants ("Long noncoding RNA" - lncRNA) La découverte récente de très nombreux lncRNA associés à des complexes spécifiques de modification de la chromatine tels que PRC2 ("Polycomb Repressive Complex 2") qui médie la triméthylation H3K27me3, suggère des rôles importants de nombreux lncRNA dans la régulation des états de la chromatine.
Source : K. Morris
Source : adapté de Tsai et al. (2010)
En recrutant ces protéines, HOTAIR dirige leur action de remodelage. Il en résulte une modification de la triméthylation de la chromatine (triméthylation H3K27me3 et perte de la méthylation H3K4 activante). Un lncRNA peut donc servir d'échafaudage pour des enzymes de modification des histones afin de contrôler la transcription de gènes cibles. |
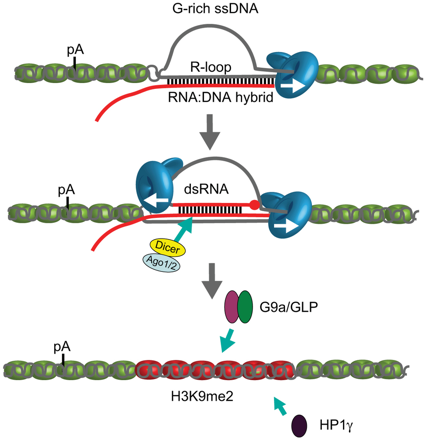
c. Rôle possible des boucles R dans la terminaison de la transcription Au cours de la transcription, la molécule d'ARN en cours de biosynthèse sort de l'ARN polymérase, ce qui rompt son appariement avec les bases complémentaires du brin d'ADN matrice. Cependant, l'ARN naissant est susceptible de se ré-hybrider au brin d'ADN transcrit, créant ainsi un hybride ARN-ADN stable et laissant le brin d'ADN non transcrit (qui a la même séquence que le brin d'ARN néo-synthétisé - l'uracile remplaçant la thymine) sous la forme d'un brin étendu non protégé. Cette structure, appelée boucle de type R ("R-loop" - triple brins - bulle de transcription), peut avoir une longueur de plus de 1000 paires de bases pour des gènes fortement transcrits. La formation de boucles de type R est donc une conséquence naturelle du processus de transcription. Les boucles de type R sont des intermédiaires clés de processus cellulaires spécifiques tels que la réplication du plasmide de Escherichia coli (ColE1), la réplication de l'ADN mitochondrial ou la commutation de classe d'immunoglobulines (recombinaison VDJ). Les boucles de type R ont cependant été considérées comme de rares sous-produits de la transcription avec des effets potentiellement néfastes sur l'intégrité du génome en raison de la fragilité du brin d'ADN transcrit qui est déplacé. Cependant, les boucles de type R ont aussi des effets bénéfiques puisque l'on observe leur formation généralisée sur des ilots CpG de promoteurs de certains gènes de l'homme. De plus, les éléments de terminaison riches en G ("G-rich ssDNA") sont enrichis en boucles de type R. Ces éléments de terminaison facilitent la pause de Pol II avant une terminaison efficace. Un lien a été établi entre les boucles de type R et la diméthylation H3K9me2 médiée par les ARN interférents au niveau des sites de pause de la terminaison dans des gènes de mammifères codant des protéines. En effet, les gènes de mammifères qui possèdent des éléments de pause en aval de leur signal fonctionnel de poly-adénylation forment des boucles de type R au niveau des sites de terminaison. Cela facilite la synthèse d'un transcrit anti-sens qui s'hybride avec le transcrit sens pour former un ARN double-brin ("double-strand RNA" - dsRNA). Cela déclenche le recrutement de DICER, d'AGO1 et d'AGO2.
Source : Skourti-Stathaki et al. (2014) Figure ci-dessus
L'histone lysine méthyl-transférase G9a (qui forme un complexe hétérotrimérique avec GLP) est à son tour recrutée : elle catalyse la formation d'une marque de répression de la transcription H3K9me2. La protéine 1γ de l'hétérochromatine ("Heterochromatin Protein 1γ" - HP1γ) est également recrutée, ce qui renforce la pause de Pol II avant une terminaison efficace de la transcription. On peut donc supposer que les boucles de type R jouent un rôle dans l'architecture de la chromatine qui permet de définir la région de terminaison pour un certains gènes de mammifères. |
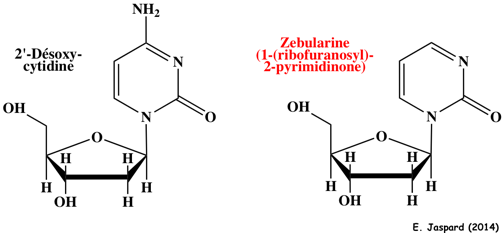
10. Méthodes pour étudier les modifications épigénétiques a. En ce qui concerne l'analyse des modifications post-traductionelles des histones et la position des acides aminés modifiés, la technique de choix est la spectromètrie de masse. b. Il n'y a pas de méthylation quand la zebularine est incorporée dans l'ADN car c'est un inhibiteur des ADN méthyltransférases.
La zebularine induit :
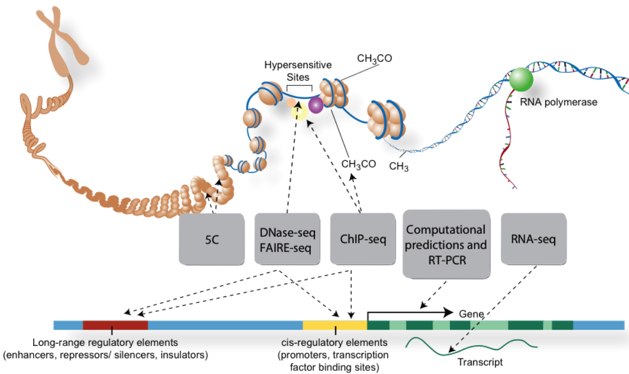
Le complexe intermédiaire covalent entre l'enzyme et l'ADN est réversible (différant ainsi des 5-fluorodésoxy)cytidines). La zebularine est aussi un inhibiteur (analogue de l'état de transition) de la cytidine désaminase. c. Figure ci-dessous : techniques de traitement des acides nucléiques avant séquençage pour l'analyse de parties spécifiques des génomes.
Source : ENCODE Voir un cours sur les nouvelles technologie de séquençage à très haut débit (NGS). |
| Eléments du génome cartographiées | Techniques utilisées |
| Sites de fixation des facteurs de transcription | ChIP-seq / DNase-seq |
| Structure de la chromatine | DNase-seq / FAIRE-seq / Histone ChIP-seq / MNase-seq |
| Sites de méthylation de l'ADN | RRBS |
|
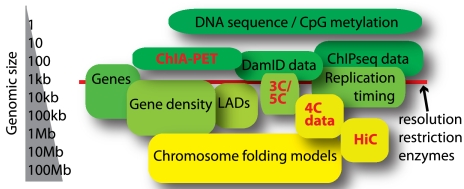
Le développement de nouvelles technologies permet l'étude du chromosome interactome et des interactions chromatine-chromatine à longue distance in vivo :
|
Source : de Wit & de Laat (2012) |
|
Définitions des acronymes de ces nouvelles technologies
ChIPBase : base de données et plate-forme pour le décodage des cartes de liaison, des facteurs de transcription, des profils d'expression, de la régulation de la transcription de longs ARN non codants ("long non-coding RNAs" : lncRNAs, lincRNAs), de miARN et autres ARN non codant (snoRNAs, tRNAs, snRNAs, ...) et des gènes codant des protéines. |
|
11. Quelques abréviations et noms de protéines impliquées dans l'épigénétique
Source : Wikipédia
|
| Abréviations | Noms |
|
Trx : Trithorax Mnn1 : Menin1 Trr : Trithorax-related Wds : Will die slowly Dpy30 : Dumpy-like 30 E(z) : Enhancer of zeste Wdr5 : WD repeat domain 5 PHF1/19 : PHD finger protein 1/19 EZH1/2 : Enhancer of zeste homolog 1/2 EED : Embryonic ectoderm development Su(var)3-9 : Suppressor of variegation 3-9 Jarid2 : Jumonji, AT rich interactive domain 2 WHSC1 : Wolf-Hirschhorn syndrome candidate 1 Rbbp4/5/7 : Retinoblastoma binding protein 4/5/7 AF17 : ALL1-fused gene from chromosome 17 Pcl : Polycomb-like Esc : Extra sexcombs HCF1 : Host Cell factor1 Escl : Extra sexcombs-like AEBP2 : AE binding protein 2 PA1 : PTIP associated factor 1 Su(z)12 : Suppressor of zeste 12 Dot1 : Disruptor of Telomeric silencing 1 NURF55 : nucleosome remodeling factor 55 SUV39H1/2 : Suppressor of variegation 3-9 homolog 1/2 MTF2 : Metal response element binding transcription factor 2 TRRAP : Transformation/transcription domain-associated protein Cps25 : 30, 35, 40 and 60, Compass subunit 25, 30, 40, 40 and 60 UTX : Ubiquitously-transcribed TPR protein on the X chromosome PTIP : PAX interacting (with transcription-activation domain) protein 1 CXXC1 : CXXC finger protein 1 dWdr82 : dWD repeat domain 82 HP1 : Heterochromatin protein 1 SETDB1 : SET domain, bifurcated 1 CAF1 : chromatin assembly factor 1 Ash2 : absent, small, or homeotic discs 2 NCOA6 : Nuclear receptor coactivator 6 MLL1/2/3/4 : Mixed-lineage Leukemia 1/2/3/4 COMPASS : Complex proteins associated with Set1 KMT : Lysine Methyl Transferase |
ASH1L : ASH1-like protein ATAD2 : ATPase family AAA domain-containing protein 2 ATAT1 : α-tubulin acetyltransferase 1 BAZ2A : bromodomain adjacent to zinc finger domain protein 2A BPTF : bromodomain PHD finger transcription factor BRD1 : bromodomain containing protein 1 BRDT : bromodomain testis-specific protein BRPF1 : bromodomain and PHD finger-containing protein 1 BRWD1 : bromodomain and WD repeat-containing protein 1 CECR2 : cat eye syndrome chromosome region candidate protein 2 CLOCK : circadian locomoter output cycles kaput protein CREBBP : CREB binding protein DOT1L : DOT1-like protein EHMT1 : euchromatic histone lysine N-methyltransferase 1 ELP3 : elongator complex protein 3 EP300 : E1A binding protein p300 EZH1 : histone lysine N-methyltransferase EZH1 GTF3C4 : general transcription factor 3C polypeptide 4 HAT : histone acetylase HDAC : histone deacetylase JARID2 : Jumonji/ARID domain-containing protein 2 JMJD1C : Jumonji domain-containing protein 1C KAT2A : lysine acetyltransferase 2A KDM : lysine demethylase KDM1A : lysine-specific histone demethylase 1A L3MBTL : lethal 3 MBT-like protein 1 MBTD1 : MBT domain-containing protein 1 MDS1 : myelodysplasia syndrome 1 MINA : MYC-induced nuclear antigen MLL : mixed lineage leukaemia MYST1 : histone acetyltransferase MYST1 NCOA1 : nuclear receptor co-activator 1 NO66 : nucleolar protein 66 NSD1 : nuclear receptor binding SET domain protein 1 PBRM1 : protein polybromo 1 PHF2 : PHD finger protein 2 PHIP : pleckstrin homology domain interacting protein PMT : protein methyltransferase PRDM1 : PR domain-containing protein 1 PRMT1 : protein arginine methyltransferase 1 SETD1A : SET domain containing protein 1A SETD2 : SET domain-containing protein 2 SETMAR : SET domain and mariner transposase fusion gene SFMBT1 : SCM-like with four MBT domains protein 1 SIRT1 : sirtuin 1 SMARCA2 : SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily A member 2 SMYD1 : SET and MYND domain-containing protein SP100 : nuclear antigen SP100 SP110 : nuclear body protein SP110 SP140 : nuclear body protein SP140 SP140L : nuclear body protein SP140-like protein SUV39H1 : suppressor of variegation 3–9 homolog 1 SUV420H1 : suppressor of variegation 4–20 homolog 1 TAF1 : TBP-associated factor 1 TAF1L : TAF1-like protein UTY : ubiquitously transcribed Y chromosome tetratricopeptide repeat protein WHSC1 : Wolf–Hirschhorn syndrome candidate 1 protein WHSC1L1 : WHSC1-like protein ZMYND8 : zinc finger MYND domain-containing protein 8 |
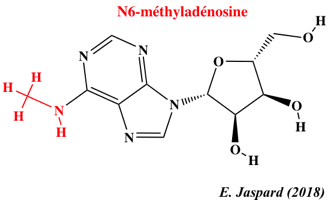
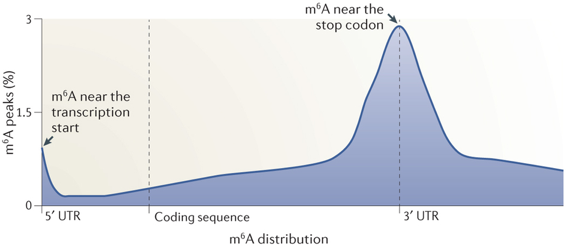
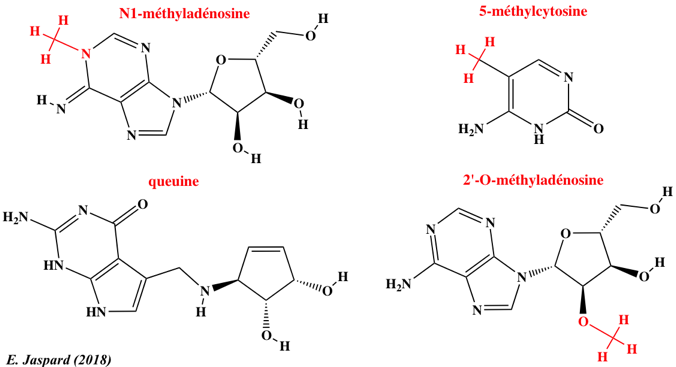
12. L'épitranscriptome : N6-méthyl-adénosine et autres nucléosides modifiés Les nouvelles technologies de séquençage ont permis d'établir que la méthylation du transcriptome (épitranscriptome) est un processus quasi général au même titre que la méthylation du génome (épigénome). L'immunoprécipitation des ARN méthylés, suivie d'une technique de séquençage à haut débit ("Methylated RNA immunoprecipitation followed by sequencing" - MeRIP-Seq) permet d'obtenir le profil de la N6-méthyl-adénosine (m6A) à l'échelle du transcriptome.
Les données actuelles de l'épitranscriptome permettent d'en décrire quelques caractéristiques :
Source : Meyer & Jaffrey (2014)
Les longs ARN non codants sont aussi sujets à la méthylation m6A. Exemples : "Long ncRNAs X inactive specific transcript" (XIST) et "HOX transcript antisense RNA" (HOTAIR - voir ci-dessus). Comme pour l'épigénome, il existe :
Outre le développement relativement récent d'anticorps anti-m6A (1988), des techniques ont été développées pour l'étude de la méthylation m6A :
Exemples de modifications d'autres nucléosides Près de 200 modifications chimiques des molécules d'ARN sont recensées. Deux technologies dominent actuellement le séquençage à lecture longue ("long-read sequencing technologies") :
RMBase : base de données qui regroupe les données de séquençage d'épitranscriptomes pour l'analyse des modifications post-transcriptionnelles des ARN. |
| 13. Liens Internet et références bibliographiques |
|
Human Epigenome Project MethBase: base de données du méthylome The Human DNA Methylome - San Diego Epigenome Center Chromatin Database Long Noncoding RNA Database Programme "Epigram" DNAmod : base de données de modifications chimiques de l'ADN RMbase : base de données de séquençage d'épitranscriptomes - analyse des modifications post-transcriptionnelles des ARN |
|
|
Richmond, Finch, Rushton, Rhodes and Klug (1984) "The structure of the nucleosome core particle at 7Å resolution" Nature 311, 532-37 Fan & Roberts (2006) "Complex of linker histone H5 with the nucleosome and its implications for chromatin packing" PNAS 103, 8384-8389 Shogren-Knaak et al. (2006) "Histone H4-K16 acetylation controls chromatin structure and protein interactions "Science 311, 844-847 Arya & Schlick (2006 ) "Role of histone tails in chromatin folding revealed by a mesoscopic oligonucleosome model" PNAS 103, 16236–16241 |
|
|
Taverna et al. (2007) "How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers" Nat. Struc. Mol. Biol. 14, 1025-1040 Graff & Mansuy (2008) "Epigenetic codes in cognition and behaviour" Behav. Brain Res. 192, 70-87 Snijders et al. (2008) "Characterization of post-translational modifications of the linker histones H1 and H5 from chicken erythrocytes using mass spectrometry" J. Proteome Res. 7, 4326-4335 Kireev et al. (2008) "In vivo immunogold labeling confirms large-scale chromatin folding motifs" Nat. Methods 5, 311-313 Park (2009) "ChIP–seq: advantages and challenges of a maturing technology" Nat. Rev. Genet. 10, 669-680 Hon et al. (2009) "Predictive chromatin signatures in the mammalian genome" Hum. Mol. Genet. 18, R195-201 |
|
|
Chandrima Das et al. (2010) "The histone shuffle: histone chaperones in an energetic dance" Trends Biochem. Sci. 35, 476-489 Woodcock & Ghosh (2010) "Chromatin higher-order structure and dynamics" Cold Spring Harb. Perspect. Biol. 2, a000596 Tsai et al. (2010) "Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes" Science 329, 689-693 Gaspar-Maia et al. (2011) "Open chromatin in pluripotency and reprogramming" Nat. Rev. Mol. Cell Biol. 12, 36-47 Bannister & Kouzarides (2011) "Regulation of chromatin by histone modifications" Cell Res. 21, 381-395 |
|
|
Cuylen & Haering (2011) "Deciphering condensin action during chromosome segregation" Trends Cell Biol. 21, 552-559 Nguyen & Zhang (2011) "The diverse functions of Dot1 and H3K79 methylation" Genes Dev. 25, 1345-1358 Khare et al. (2011) "HIstome: a relational knowledgebase of human histone proteins and histone modifying enzymes" Nuc. Acids Res. 40(D1): D337-D342 Deaton & Bird (2011) "CpG islands and the regulation of transcription" Genes Dev. 25, 1010 - 1022 |
|
|
Chu et al. (2011) "Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions" Mol. Cell. 44, 667-678 Arrowsmith et al. (2012) "Epigenetic protein families: a new frontier for drug discovery" Nat. Rev. Drug Discov. 11, 384-400 Luger et al. (2012) "New insights into nucleosome and chromatin structure: an ordered state or a disordered affair ?" Nat. Rev. Mol. Cell Biol. 13, 436–447 Jakovcevski & Akbarian (2012) "Epigenetic mechanisms in neurological disease" Nat. Medicine 18, 1194–1204 Aguilera & Garcia-Muse (2012) "R Loops: From Transcription Byproducts to Threats to Genome Stability" Mol. Cell 46, 115-124 |
|
|
Patel et al. (2013) "Readout of Epigenetic Modifications" Ann. Rev. Biochem. 82, 81-118 Kohli & Zhang (2013) "TET enzymes, TDG and the dynamics of DNA demethylation" Nature 502, 472-479 Stasevich et al. (2014) "Regulation of RNA polymerase II activation by histone acetylation in single living cells" Nature doi:10.1038 Wu & Zhang (2014) "Reversing DNA Methylation: Mechanisms, Genomics, and Biological Functions" Cell 156, 45-68 Matzke & Mosher (2014) "RNA-directed DNA methylation: an epigenetic pathway of increasing complexity" Nature Rev. Genet. 15, 394-408 Skourti-Stathaki et al. (2014) "R-loops induce repressive chromatin marks over mammalian gene terminators" Nature doi:10.1038 |
|
|
Muller & Muller (2014) "Structural biology: Enzyme–chromatin complex visualized" Nature 514, 572–573 McGinty et al. (2014) "Crystal structure of the PRC1 ubiquitylation module bound to the nucleosome" Nature 514, 591-596 |
|
|
Meyer et al. (2012) "Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons" Cell 149, 1635-1646 Dominissini et al. (2012) "Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq" Nature 485, 201-206 Meyer & Jaffrey (2014) "The dynamic epitranscriptome: N6-methyladenosine and gene expression control" Nat. Rev. Mol. Cell Biol. 15, 313-326 Basnet et al. (2014) "Tyrosine phosphorylation of histone H2A by CK2 regulates transcriptional elongation" Nature doi: 10.1038 |
|
|
Roadmap Epigenomics Consortium et al. (2015) "Integrative analysis of 111 reference human epigenomes" Nature 518, 317–330 Whitaker et al. (2015) "Predicting the human epigenome from DNA motifs" Nature Met. 12, 265 - 272 Boettiger et al. (2016) "Super-resolution imaging reveals distinct chromatin folding for different epigenetic states" Nature Watson & Tsai (2017) "Molecular biology: Local metabolites linked to memory" Nature 46, 361 - 362 |
|
|
Huang et al. (2018) "Lysine benzoylation is a histone mark regulated by SIRT2" Nat. Comm. 9, 3374 Lorna et al. (2019) "Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3" Nature Cervantes & Sassone-Corsi (2019) "Modification of histone proteins by serotonin in the nucleus" Nature 567, 464 - 465 Cavalli & Heard (2019) "Advances in epigenetics link genetics to the environment and disease" Nature 571, 489 - 499 Zhang et al. (2019) "Metabolic regulation of gene expression by histone lactylation" Nature 574, 575 - 580 |
|
|
Bhat et al. (2021) "Epigenetics and beyond: targeting writers of protein lysine methylation to treat disease" Nat. Rev. Drug Discov. 20, 265 - 286 Grand et al. (2021) "BANP opens chromatin and activates CpG-island-regulated genes" Nature doi: 10.1038/s41586-021-03689-8 Minnoye et al. (2021) "Chromatin accessibility profiling methods" Nat. Rev. Meth. Primers 1, Art. number 10 Lucas & Novoa (2023) "Long-read sequencing in the era of epigenomics and epitranscriptomics" Nat. Methods 20, 25 - 29 |
![]()
{kind=link}