
Les marqueurs de séquence exprimée ou "EST" ("expressed sequence tags") |
| Tweet |
|
|
1. Introduction 2. Principe de l'obtention d'EST 3. Les biais
4. La banque "dbEST" 5. Le regroupement des EST
|
6. L'assemblage des EST et des transcrits
7. Exemples de résultats issus de l'analyse d'EST 9. Liens Internet et références bibliographiques |
|
1. Introduction Le génome des Eucaryotes ne contient qu'une proportion faible de séquences dites codantes : séquences d'ADN transcrites en ARN messagers, eux-mêmes traduits en protéines. Il existe une différence de niveau de transcription des gènes selon les tissus et dans le temps (différence de transcription spatio-temporelle). Dans un tissu donné, environ 15.000 gènes sont transcrits:
Le taux de transcription des gènes, c'est-à-dire la quantité d'ARN (messagers ou non codants) est trés variable. En conséquence, le séquençage d'ADN complémentaire ou ADNc consiste à caractériser l'ensemble des ARN messagers (qui auront été préalablement rétro-transcrits en ADNc) synthétisés dans une cellule. |
|
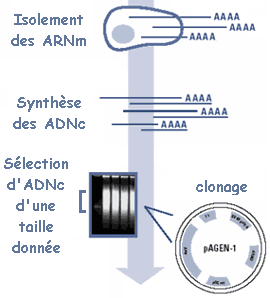
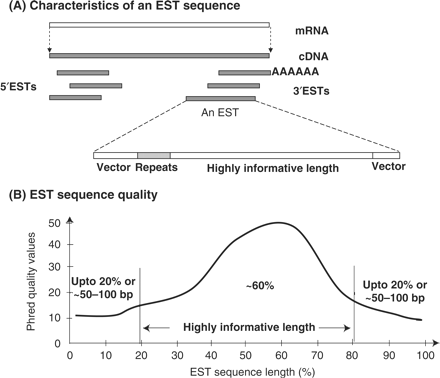
2. Principe de l'obtention d'EST a. La première étape est la construction d'une banque d'ADNc : ceux-ci sont clonés de sorte que l'on obtienne une collection de clones indépendants. Pour celà, les ADNc sont insérés dans un même type de vecteur, choisi en fonction du type de banque que l'on veut construire.
L'énorme intérêt est que toutes les séquences en amont et en aval des ADNc (séquences d'ADN du vecteur) sont identiques pour tous les clones : on utilise les mêmes jeux d'amorces pour le séquençage de tous les clones, ce qui permet l'automatisation et donc un séquençage à grande échelle. b. Pour chaque clone, quelques centaines de nucléotides (200 à 700) sont séquencés une seule fois ("single pass") à chaque extrémité de l'ADNc inséré. L'information peut donc n'être que partielle par rapport à la taille de certains ADNc (qui peut atteindre plusieurs milliers de nucléotides), mais elle est suffisante pour caractériser de manière univoque chaque clone. Ces séquences partielles d'ADNc sont appelées marqueurs de séquence exprimée ou "EST" : "expressed sequence tags". Le séquençage s'effectuant à partir des 2 extrémités, on génère 2 types d'EST :
|
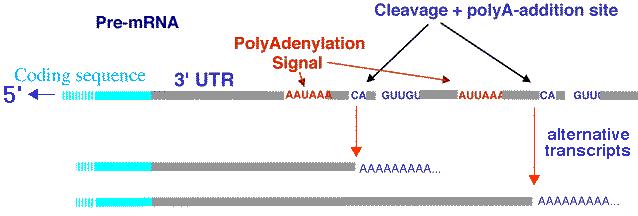
5' EST Les 5' EST sont obtenus à partir de la région des transcrits qui, le plus souvent, est celle qui code une protéine. Cette région est davantage conservée entre différentes espèces et ne changent pas beaucoup au sein d'une famille de gènes. 3' EST Les 3' EST ont une probabilité plus élevée de correspondre à une région non-codante ou non traduite ("UnTranslated Regions" - UTR) et sont caractérisés par une plus faible conservation entre différentes espèces. Par ailleurs, une étude des transcrits de 10 chromosomes humains a montré que prés de la moitié sont non polyadénylés [poly(A)-] :
Cheng et al. (2005) "Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution" Science 308, 1149 - 1154 Enfin, la polyadénylation alternative génère différents transcrits à partir d'un même gène (schéma ci-dessous).
Source : D. Gautheret - INSERM ERM206 Polyadénylation chez les procaryotes : voir Sarkar (1997) "Polyadenylation of mRNA in prokaryotes" Annual Rev. Biochem. 66, 173-197 |
Quelques applications des EST :
|
Avantages des EST :
Source : Nagaraj et al. (2007) Inconvénients des EST :
|
|
3. Les biais a. La normalisation des banques d'ADNc (Soares et al., 1994) La différence de niveau de transcription des gènes est régulée par de nombreux systèmes de transduction du signal. La différence de niveau de transcription d'un gène peut varier de 1 copie à 25.0000 copies par cellule selon le contexte. Les gènes peuvent être classés en 3 catégories sur la base du nombre de copies de leurs ARN messagers à un moment donné :
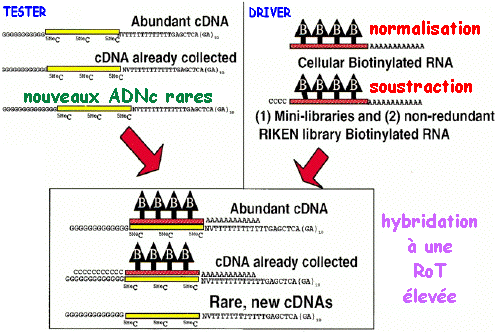
Cette gamme extrèmement étendue de niveau de transcription d'un gène à un autre rend difficile l'analyse de banques d'ADNc en particulier si la détermination de séquences est la stratégie choisie pour la découverte de gènes. La normalisation est un processus complexe au cours duquel le nombre de copies de tous les ADNc d'une banque "primaire" est égalisé afin que tous les transcrit soit présents de façon équivalente dans la banque normalisée. La technique de normalisation s'appuie sur une propriété cinétique de l'hybridation entre molécules d'acides nucléiques. La grande difficulté est de sélectionner les molécules simple brin. L'hybridation entre acides nucléiques dépend de nombreux paramètres physico-chimiques. En particulier, la probabilité d'hybridation entre des séquences complémentaires augmente avec :
Pour tenir compte de ces 2 facteurs, on définit le produit (concentration x temps) appelé :
En pratique, le nombre de copies des gènes fortement et modérément exprimés est donc réduit (en moyenne d'un facteur 100) à celui des gènes rares dans la banque normalisée. En conséquence :
|
|
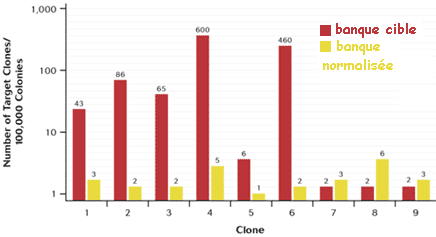
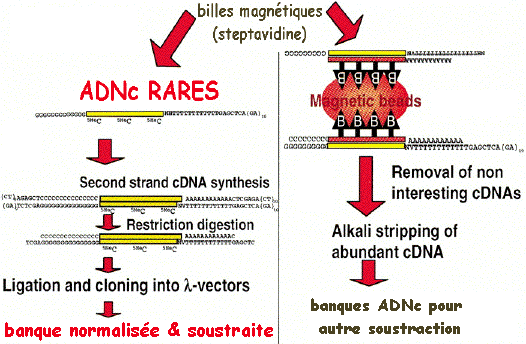
c. Exemple de protocole pour la préparation d'une banque normalisée et soustraite en une seule étape (Carninci et al., 2000) La stratégie repose sur :
La normalisation et la soustraction sont effectuées en une seule étape.
|
|
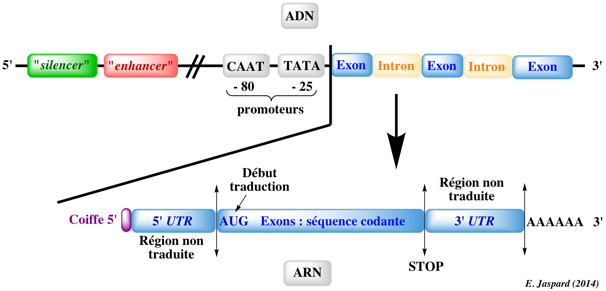
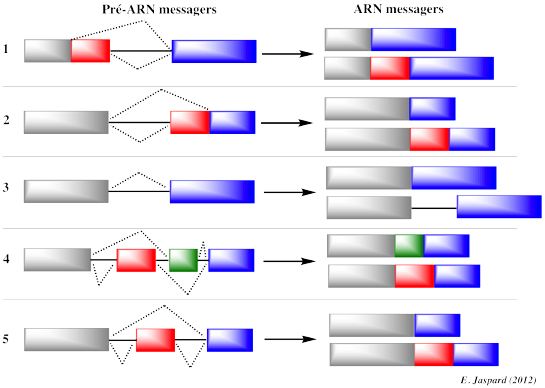
Les génes sont transcrits sous forme d'ARN messagers pré-matures (synonymes : transcrits primaires ou pré-ARNm) qui contiennent des introns (séquences de l'ARN pré-mature non retenues dans la séquence finale de l'ARN messager qui code la protéine) et des exons qui sont assemblés selon différentes combinaisons (épissage alternatif). Les introns ont des tailles extrêmement variables : de plusieurs centaines de nucléotides à plusieurs centaines de milliers de nucléotides. L'épissage alternatif est le processus qui permet à un même gène de générer différents transcrits selon la combinaison des exons qui formeront l'ARN messager mature. L'épissage est effectué par deux réactions de trans-estérification au sein de complexes appelés spliceosomes formés, entre autres, de 5 particules ribonucléoprotéiques appelées SnRNP ("Small nuclear RiboNucleoProtein"). Ce sont des protéines associées à des petits ARN nucléaires ("small nuclear RNA" - snRNA) riches en uracile (U1, U2, U4, U5 et U6). Figure ci-dessous : Les 5 types d'épissage alternatif
|
Les répercutions de l'épissage alternatif sur les EST que l'on peut obtenir sont les suivantes :
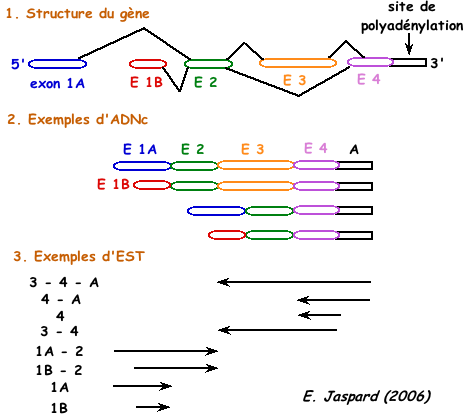
Figure adaptée de "Précis de génomique", Gibson & Muse (2004) Puisque les séquences des EST ne correspondent qu'aux extrémités 3' ou 5' des ADNc, des séquences distinctes d'EST issues d'un même gène peuvent être interprétées dans un premier temps comme issues de gènes différents. La comparaison des EST et des séquences d'ADN génomique permet de lever cette ambiguïté et d'associer différentes EST à un gène unique. |
|
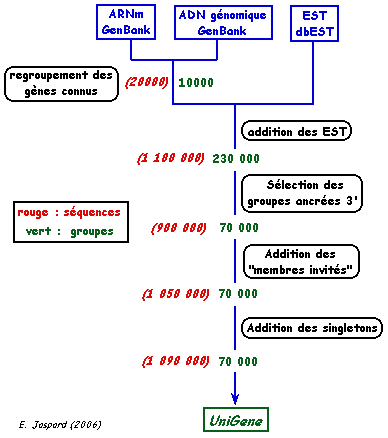
La banque "dbEST" du NCBI (créée en 1993) contient 74 millions d'EST (juillet 2018) obtenues par différents projets de séquençage. Des banques diminuent l'extrème redondance des séquences d'EST en regroupant les séquences correspondant au même gène : UniGene La banque "dbGSS" ("database of Genome Survey Sequence") est semblable à dbEST, mais les séquences sont d'origine génomique et non issues d'ADNc (plus de 35 millions de GSS - Janvier 2013). |
| organisme | nombre d'EST |
| Homo sapiens | 8 705 000 |
| Arabidopsis thaliana | 1 530 000 - (1 046 000) |
| Zea mays (mais) | 2 019 000 |
| Oryza sativa (riz) | 1 253 000 |
| Triticum aestivum (wheat) | 1 286 000 |
|
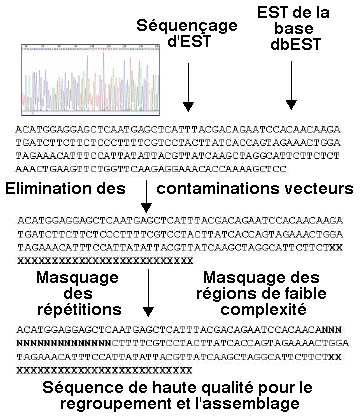
5. Le regroupement des EST Le regroupement ("clustering") des EST nécessite un pré-traitement des séquences peu ou pas spécifiques.
Source : MUBRI Bioinformatics Group Ces séquences sont :
Exemples de séquences de faible complexité :
Exemple de programme qui analyse ce type de régions : "SEG" (partie "Algorithm parameters" de BLAST). |
|
b. Les groupes "UniGene" - NCBI UniGene est un système de regroupement automatique des séquences (et donc des EST) de la base de données GenBank dans différents ensembles non redondants de groupes ou "clusters". Il existe des groupes "UniGene" pour plusieurs dizaines d'espèces animales et végétales. On constate que les chiffres n'augmentent plus beaucoup depuis 2010. Est-ce dû à l'apparition des nouvelles techniques de séquençage à trés haut débit ("NGST") qui permettent une étude du transcriptome à bien plus grande échelle et beaucoup plus précisément (au nucléotide près), avec des conditions expérimentales moins complexes et pour un coût moindre ? En d'autres termes : les approches EST et puces à ADN sont-elles appelées à disparaître au profit d'approche telles que "RNA seq" ou "MPSS" ? Chaque groupe "UniGene" contient :
"ProtEST" ("Protein/EST Alignments") est un outil qui propose des alignements (déjà générés par BLAST) entre les séquences de protéines d'organismes modèles et la traduction selon les 6 phases de lectures de séquences nucléotidiques issues de "UniGene". La collection "UniGene" a été utilisée comme source de séquences uniques pour la fabrication de puces à ADN. |
|
c. Construction itérative d'un groupe "UniGene" sur la base de transcrits (ARNm) Les séquences sont d'abord masquées par le programme DUST (NCBI). Seules les EST d'au moins 100 paires de bases significatives (et avec un trés petit nombre de "N") sont retenues pour être intégrées à un groupe "UniGene". 1ère analyse :
2ème analyse :
Voir la répartition finale des groupes "UniGene" pour Arabidopsis thaliana L'ensemble des groupes d'EST ainsi construits est comparé à l'ensemble des groupes d'EST construits la semaine précédente et re-numérotés. Puisque le nombre de séquences constitutives d'un groupe peut changer et que l'identifiant d'un groupe peut disparaître (par exemple quand 2 groupes fusionnent / voir le groupe "UniGene" At.49097), il est préférable d'utiliser le numéro d'accession Genbank d'une séquence. |
d. Exemple d'un groupe "UniGene" Groupe "UniGene" At.49098 - Arabidopsis thaliana
RAFL : acronyme pour "RIKEN Arabidopsis Full-Length cDNAs" |
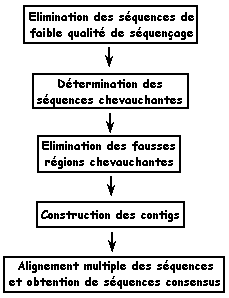
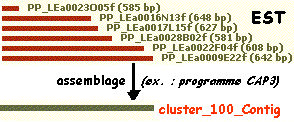
6. L'assemblage des EST et des transcrits a. Les contig (définition originale donnée par Staden, 1979)
La comparaison des séquences permet d'aligner les parties qui se recouvrent partiellement ou"chevauchantes". Les séquences chevauchantes peuvent être assemblées en enchaînements plus grands que l'on appelle des contigs. Cette opération d'assemblage est effectuée par des programmes informatiques tels que :
|
|
b. La reconstitution de la structure la plus probable d'un gène Elle nécessite d'isoler des ADNc pleine longueur (criblage par hybridation de banque de haute qualité) et de connaître la séquence du génome. La position réelle de l'extrémité 5' peut être déterminée par des techniques telles que l'extension d'amorce ou la protection contre la RNase. Des programmes informatiques tentent de prédire le ou les produits de l'unité de transcription. a. Il faut prédire les séquence signal de la transcription et de la traduction :
b. Il faut prédire les sites d'épissage corrects des exons pour identifier les différents transcrits alternatifs :
Parmi ces programmes, on peut citer, par exemple : |
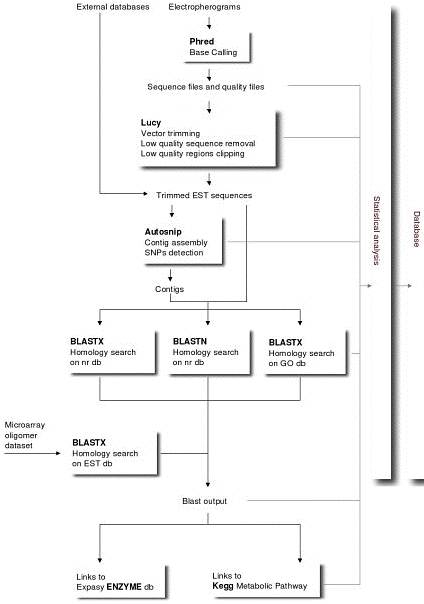
Les "pipeline" Ce sont des suites logicielles qui permettent le traitement d'un trés grand nombre de données de manière automatisées. Les données de bases sont traitées par un premier logiciel puis les résultats sont soumis à un autre logiciel et ainsi de suite jusqu'à l'obtention d'une information riche en renseignements pour le biologiste. Ci-dessous, une description de l'ensemble du processus du traitement d'EST de la pêche :
Source : ESTree db
|
| Les analyses comparatives suggèraient donc à cette époque que : | ||
| les gènes impliqués dans : |
|
sont plus actifs que ceux des feuilles exemptes de PPV. |
| un certain nombre de produits de transcription, dont : |
|
ne sont très fréquents que dans les feuilles de pêcher infectées par le PPV. |
| Les résultats de cette étude ont aidé à mieux comprendre les mécanismes moléculaires associés à la sensibilité à la sharka chez le Prunus persica et ont facilité le développement de nouvelles stratégies de lutte contre le PPV. | ||
|
b. Melotto et al. (2005) "Comparative bioinformatic analysis of genes expressed in common bean (Phaseolus vulgaris L.) seedlings" Genome 48, 562 - 570 Découverte de marqueurs moléculaires pour l'étiquetage et la cartographie de gènes exprimés chez le haricot.
|
| 8. Liens Internet et références bibliographiques |
|
"Précis de génomique", Gibson & Muse (2004) Adams et al. (1991) "Complementary DNA sequencing: expressed sequence tags and human genome project" Science 252, 1651 - 1656 |
|
|
Ganeteg et al. (2004) "Is Each Light-Harvesting Complex Protein Important for Plant Fitness ?" Plant Physiol., 134, 502 - 509 Nagaraj et al. (2007) "A hitchhiker's guide to expressed sequence tag (EST) analysis" Brief Bioinform. 8, 6 - 21 Boguski et al. (1993) "dbEST-database for expressed sequence tags" Nat Genet. 4, 332 - 333 Boguski et al. (1993) "dbEST-database for expressed sequence tags" Nat. Genet. 4, 332 - 333 Pontius et al. (2003) "UniGene : a unified view of the transcriptome" The NCBI Handbook - Bethesda (MD) - National Center for Biotechnology Information |
|
|
Soares et al. (1994) "Construction and characterization of a normalized cDNA library" Proc. Natl. Acad. Sci. USA 91, 9228 - 9232 Diatchenko et al. (1996) "Suppression subtractive hybridization: A method for generating differentially regulated or tissue-specific cDNA probes and libraries" Biochemistry 93, 6025 - 6030 Asamizu et al. (2000) "A large scale analysis of cDNA in Arabidopsis thaliana: generation of 12,028 non-redundant expressed sequence tags from normalized and size-selected cDNA libraries" DNA Res. 7, 175 - 180 Carninci et al. (2000) "Normalization and subtraction of cap-trapper-selected cDNAs to prepare full-length cDNA libraries for rapid discovery of new genes" Genome Res. 10, 1431 - 1432 |
|
|
Huang & Madan (1999) "CAP3: A DNA Sequence Assembly Program" Genome Res. 9, 868 - 877 "CAP3" Program (implémentation au PBIL - Lyon) Künne et al. (2005) "CR-EST: a resource for crop ESTs" Nuc. Acids Res., 33, D619 -D621 CR-EST : "The crop expressed sequence tag database" "TAIR SeqViewer Whole Genome View" Staden, R. (1979) "A strategy of DNA sequencing employing computer programs", Nucleic Acids Res. 7, 2601 - 2610 |
|
![]()

{kind=link}