Quelques méthodes d'analyse quantitative du transcriptome et du niveau de transcription des gènes (méthodes SAGE et ses dérivés - MPSS - CAGE - RNAseq) |
| Tweet |
|
|
1. Introduction 2. La méthode SAGE et ses dérivées
3. La banque de données SAGEmap et "Gene Expression Omnibus" |
4. Les autres méthodes : MPSS, CAGE, RNA-seq
5.Les frontières intron-exon ("exon-intron borders") 6. Liens Internet et références bibliographiques |
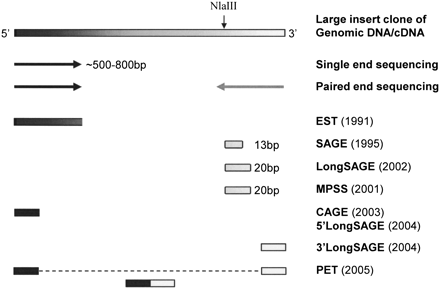
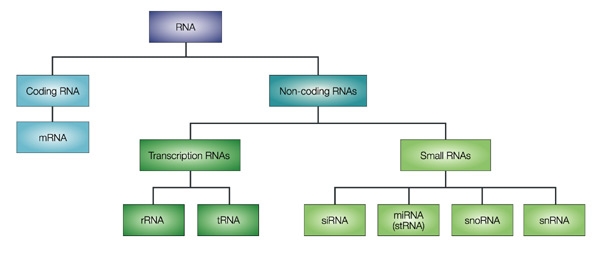
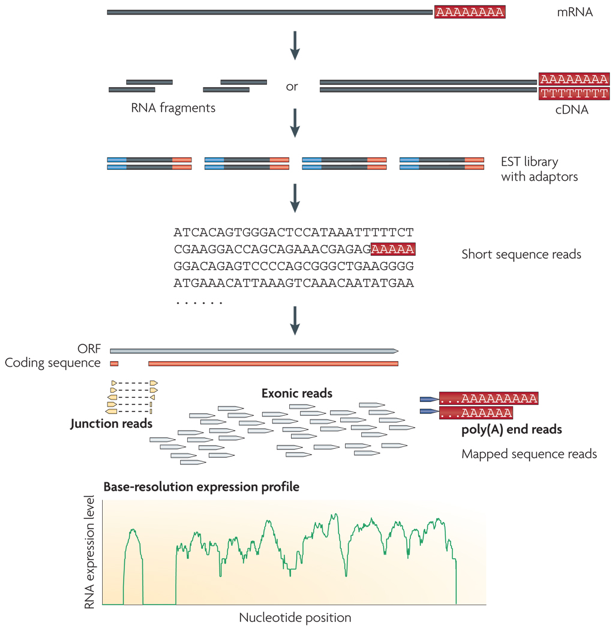
1. Introduction Les grands fragments d'ADN peuvent être analysés par séquençage complet, par séquençage à partir de l'une des deux extrémités ("single-end sequencing") ou par séquençage à partir des deux extrémités ("paired-end sequencing" - PET). La méthode des EST a été la première à générer une étiquette pour chaque fragment de cDNA séquencé.
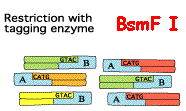
La méthode SAGE emploie une enzyme d'étiquetage (l'enzyme de restriction NlaIII qui coupe 13 pb en aval de son site de reconnaissance). Cette méthode génère des étiquettes de 13 pb de l'extrémité 3'.
Source : Fullwood et al. (2009) Les méthodes LongSAGE et MPSS emploient l'enzyme de restriction MmeI qui coupe de 18 à 20 pb en aval de son site de reconnaissance et générent donc des étiquettes de 20 pb qui peuvent être beaucoup plus spécifiquement alignées / comparées avec des génomes dits de référence. Les étiquettes issues des méthodes CAGE et 5′ LongSAGE sont obtenues à partir de l'extrémité 5′ des fragments de cDNA. Les étiquettes issues de la méthode 3′ LongSAGE sont obtenues à partir de l'extrémité 3′ des fragments de cDNA. La technologie PET ("paired-end tags") combine de manière covalente les étiquettes en 5′ et 3′ (20 - 30 pb chacune) d'un même fragment de cDNA en une seule double-étiquette. La méthode SAGE est présentée en premier car elle est l'une des plus anciennes. Cela permet de définir des termes, des principes, des démarches qui, à peu de choses près, sont identiques pour toutes les autres méthodes. Ces dernières sont en général des affinements, et/ou s'appuient sur des technologies de séquençage à trés haut débit apparues récemment. |
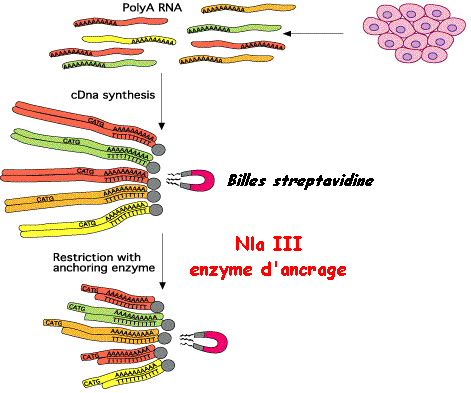
2. La méthode SAGE et ses dérivées (LongSAGE, SuperSAGE, DeepSAGE) La méthode d'analyse sérielle de l'expression des gènes ou méthode SAGE permet une analyse de la fréquence d'expression d'un ARN messager parmi les milliers de produits dans une cellule, à un moment donné. Elle se fonde sur l'analyse séquentielle d'un grand nombre de courts fragments d'ADNc, dont chacun représente la signature d'un gène. C'est une méthode de choix pour l'analyse du transcriptome, car elle permet :
La méthode SAGE diffère de l'approche par analyse d'EST car :
|
|
a. Principe de la méthode SAGE
Etape 1
Source : SAGE -"Science Park"
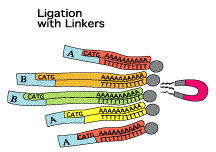
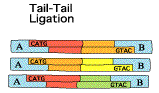
L'un des points originaux de la méthode SAGE est de dupliquer l'ensemble de ces opérations (2 prélèvements disctincts du même échantillon d'ARN messagers de départ). Le but de cette duplication est de traiter chaque échantillon avec 1 adaptateur particulier. Etape 2
Etape 3
En revanche, ils diffèrent tous à leur extrémité 3' par les 10 à 14 nucléotides spécifiques de leur étiquette. Etape 4
Etape 5
Source : SAGE - "Science Park"
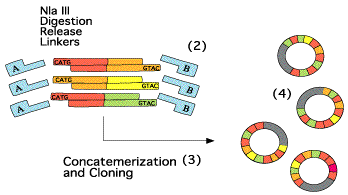
Etape 6
Exemple de concatémères :
L'analyse bioinformatique des données est effectuée avec des logiciels spécifiques qui :
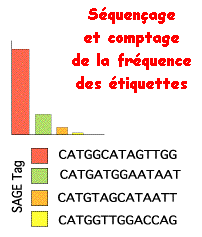
Résultat : le dénombrement des ARN messagers permet la mesure quantitative du profil d'expression dans différentes conditions physiologiques.
La comparaison avec des banques de données de séquences de génomes complets permet d'identifier les gènes mis en évidence. étiquette Nombre Nom du gène ATATTGTCAA 5 translation elongation factor 1 gamma AAATCGGAAT 2 T-complex protein 1, z-subunit ACCGCCTTCG 1 no match GCCTTGTTTA 81 rpa1 mRNA fragment for r ribosomal protein GTTAACCATC 45 ubiquitin 52-AA extension protein CCGCCGTGGG 9 SF1 protein (SF1 gene) TTTTTGTTAA 99 NADH dehydrogenase 3 (ND3) gene GCAAAACCGG 63 rpL21 GGAGCCCGCC 45 ribosomal protein L18a GCCCGCAACA 34 ribosomal protein S31 GCCGAAGTTG 50 ribosomal protein S5 homolog (M(1)15D) TAACGACCGC 4 BcDNA.GM12270 |
|
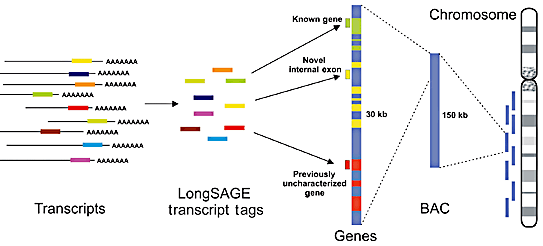
b. Méthodes LongSAGE, SuperSAGE et DeepSAGE La méthode LongSAGE est une adaptation de la méthode SAGE. Elle génère des étiquettes plus longues de 14 à 18 paires de base et un site de restriction pour l'endonuclease MmeI.
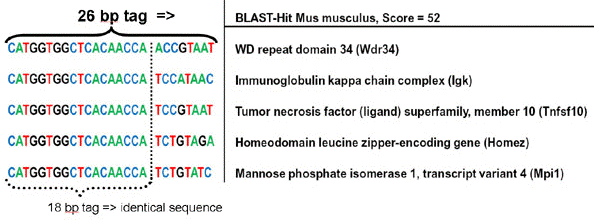
Source : Saha et al. (2002) Plus récemment encore la méthode SuperSAGE a été développée (Matsumura et al., 2003). Elle génère des étiquettes encore plus longues de 26 paires de base et un site de restriction pour l'endonuclease Ecop15I (endonucléase du phage P1). La figure ci-dessous montre l'intérêt d'étiquettes plus longues : les 18 premières bases ne permettent pas de discriminer.
Source : Genxpro Les étiquettes de de 26 paires de base sont 17.000 fois plus spécifiques que des étiquettes de 18 paires de base. Avantages de cette méthode :
Enfin, la méthode DeepSAGE combine :
300.000 étiquettes sont ainsi générées en une seule analyse ! |
|
3. La banque de données "SAGEmap" et "Gene Expression Omnibus" a. "SAGEmap" (NCBI) - (autre lien) Elle permet d'établir une relation entre des étiquettes SAGE ou des étiquettes LongSAGE (17 nucléotides) et les groupes ("clusters") "UniGene". Ces groupes contiennent des séquences qui représentent un gène unique (voir : Arabidopsis thaliana : UniGene Build #72). "SAGEmap" contient plusieurs centaines d'expériences SAGE (banques d'ADNc dans différentes conditions). b. Gene Expression Omnibus (GEO) C'est une base de données d'expression et d'abondance de molécules (ARNm, ADN génomique et protéines) et aussi un système de recherche de ces données d'expression. Les données soumises répondent à la charte de standardisation "MIAME". Les données de GEO sont issues de diverses technologies : puces à ADN, méthode SAGE et spectromètrie de masse. |
4. Les autres méthodes a. La méthode MPSS - "Massively parallel signature sequencing" - Brenner et al. (2000) Méthode liée à l'apparition des technologies de séquençage à trés haut débit qui ont révolutionné la portée et l'ampleur des études en génomique. Elle génère des étiquettes de 16 à 20 pb (en moyenne 17 pb). Cette identification est effectuée en parallèle sur des centaines de milliers de billes et environ 1 million de signatures sont obtenues par expérience. Avantages de cette technique :
Source : Buckingham S. (2003) Application à Arabidopsis
Remarque : une étude récente (Yamamoto et al., 2009) combinant la méthode "Cap-trapper" (Carninci et al., 1996) qui permet de sélectionner des transcrits entiers et la méthode MPSS a analysé en détail les promoteurs de Arabidopsis.
|
|
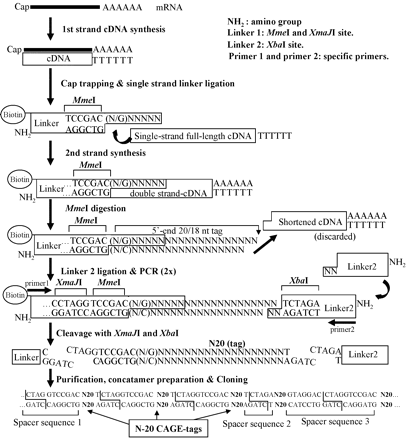
La méthode CAGE ("Cap analysis gene expression") est basée sur la méthode "Cap-trapper" (Carninci et al., 1996) qui permet de sélectionner des transcrits entiers ("full-length RNAs") qui contiennent donc entièrement leur extrémité 5'. Les ARNr et les ARNt ne sont pas retenus.
Source : Shiraki et al. (2003) Il s'ensuit un marquage à l'extrémité 5' qui introduit un site de reconnaissance d'une enzyme de restriction (MmeI). La suite de la méthode CAGE n'est autre que la méthode LongSAGE pour générer les étiquettes CAGE. La méthode CAGE produit donc des étiquettes (environ 20-21 pb) à partir du tout début de l'extrémité 5' des ARNm ("5' ends of capped transcripts"). Les étiquettes CAGE ont pour but de localiser avec exactitude les sites de démarrage de la transcription dans le génome. Cette méthode permet d'étudier la stucture des régions promotrices.
Ces régions peuvent être identifiées en comparant des séquences génomiques relativement distantes sur le plan évolutionnaire, en focalisant sur les régions conservées en amont des gènes annotés. Une version simplifiée de la méthode CAGE a été développée : le premier brin synthétisé d'ADNc est séquencé par une technologie de séquençage à trés haut débit. Elle s'affranchit donc de la synthèse du second brin d'ADNc, de l'amplification, de la ligation / digestion. Base de données : "CAGE homepage at the RIKEN Omics Science Center" |
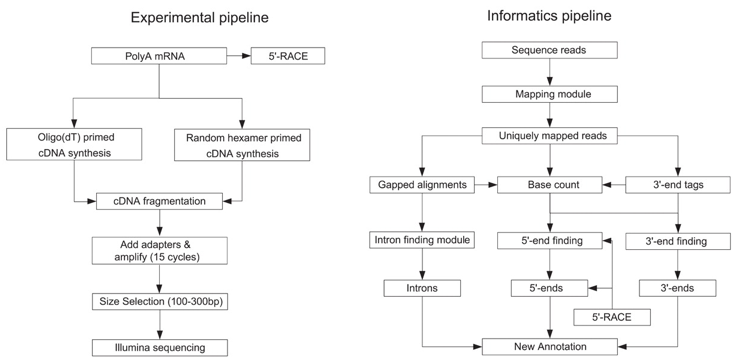
c. La méthode "RNA-seq" ou "Whole Transcriptome Shotgun Sequencing" - WTSS C'est une méthode récente (protocole général ci-dessous). De nombreuses adaptations de certaines des étapes ont trés vite été développées. Elle est aussi liée à l'apparition des technologies de séquençage à trés haut débit. Des millions de fragments ("ultra high-throughput short reads") sont générés et séquencés.
Source : Nagalakshmi et al. (2008) Principales étapes (ci-dessous) :
Source : Wang et al. (2009)
Quelques avantages de la méthode "RNA-seq" ("RNA-sequencing")
|
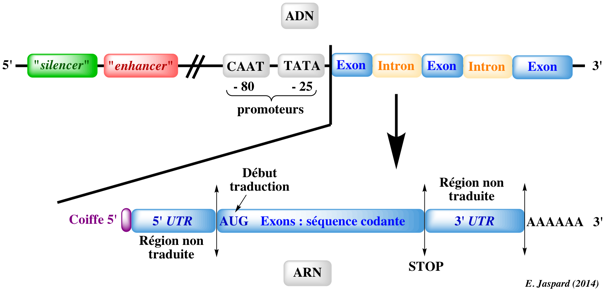
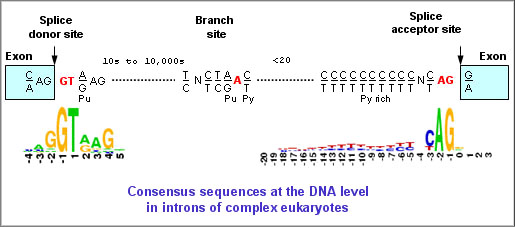
5. Les frontières intron-exon ("exon-intron borders") La plupart des introns commencent par la séquence consensus GU et finissent par la séquence consensus AG (sens 5' vers 3'). Ces séquences sont appelés respectivement "site donneur lors de l'épissage" et "site accepteur lors de l'épissage" ("splice donor site" et "splice acceptor site"). Il existe en général une région riche en nucléotides pyrimidiques (C et U) en amont du site AG. En amont de cette région se trouve le "point de branchement" ("branch point") qui contient toujours une adénine, mais qui par ailleurs est faiblement conservée. Une séquence typique est YNYYRAY où Y est une pyrimidine (C ou U), N n'importe quel nucléotide, R est une purine (G ou A) et A est l'adenine. Dans 60% des cas, l'extrémité de la séquence de l'exon situé en 5' (site donneur) est (A/C)AG et l'extrémité de la séquence de l'exon situé en 3' (site accepteur) est G (voire A).
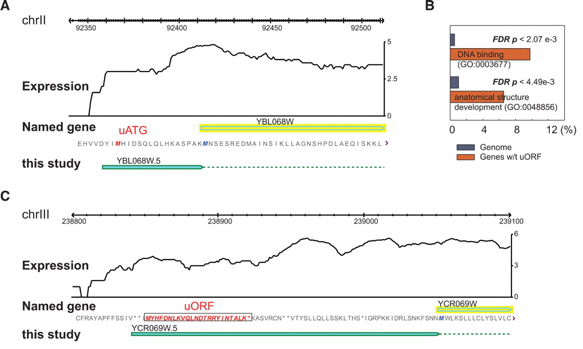
Source : "RNA sequence analysis tools" L'analyse du transcriptome de la levure prédit l'existence de telles ORF pour 6% des transcrits, en particulier en ce qui concerne les gènes codant des protéines se fixant à l'ADN. Dans la figure A ci-dessous, on notera l'échelle trés précise sur le chromosome II. Cette étude a porté également sur la transcription dans les régions intergéniques en identifiant des segments d'au moins 150 pb avec un taux d'expression significativement supérieur aux segments environnants. 204 segments de ce type (non observés par la technologie des puces à ADN) ont ainsi été mis en évidence.
Source : Nagalakshmi et al. (2008) De nombreuses variantes de la méthode "RNA-seq" d'origine ont été développées pour étudier telle ou telle partie du génome via le transcriptome. La figure ci-contre en montre un exemple :
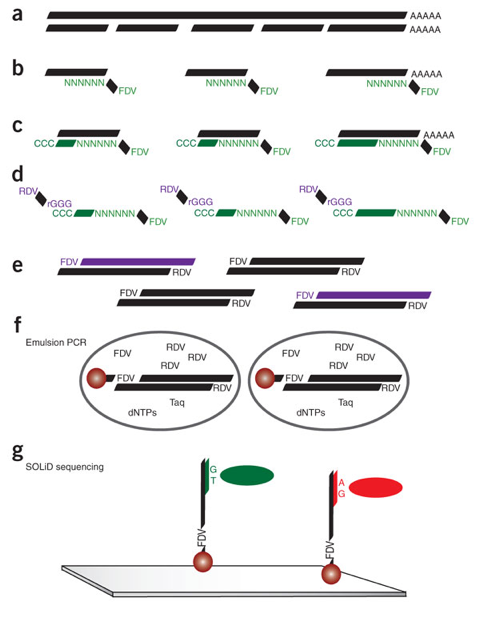
Source : Cloonan et al. (2008) (a) Fragmentation des ARN messagers. (b) Synthèse du premier brin de cDNA avec une amorce hexamère aléatoire marqué par une séquence flanquante (FDV). (c) Des cytosines sont ajoutées à chaque cDNA. (d) Une hybridation entre le cDNA (vert) et un fragment d'ARN marqué par une seconde séquence flanquante (RDV) permet d'incorporer un site de marquage RDV dans le premier brin de cDNA. (e) La banque est amplifiée par PCR avec des amorces FDV et RDV. (f) Les fragments amplifiés sont attachés à des billes (pour le séquençage à suivre) par émulsion. (g) Les billes sont covalamment fixées à un support. De courtes séquences (25 à 35 nucléotides) sont générées via la technologie "SOLID" ("Sequencing by Oligonucleotide Ligation and Detection") : le séquençage est basé sur l'amplification par émulsion et l'hybridation-ligature chimique. Il utilise une ligation avec une DNA ligase. Voir un développement de cette technique et du principe des réactions chimiques. |
Moyens bioinformatiques pour ces types de méthodes En parallèle de ces technologies massivement productives de courts fragments séquencés ("ultra high-throughput short reads") ont été développés :
Autres exemples de programmes |
| 6. Liens Internet et références bibliographiques |
|
Méthode SAGE d'origine : Velculescu et al. (1995) "Serial analysis of gene expression" Science 270, 484 - 487 / Description de modifications du protocole initial de la méthode SAGE Lash et al. (2000) "SAGEmap : a public gene expression resource" Genome Res. 7, 1051 - 1060 / Bases de données SAGEmap et MAP Viewer (NCBI) Barrett et al. (2005) "NCBI GEO : mining millions of expression profiles : database and tools" Nucleic Acids Res. 33, D562 - D566 / GEO ("Gene Expression Omnibus") |
|
|
Saha et al. (2002) "Using the transcriptome to annotate the genome" Nature Biotechnol. 20, 508 - 512 Méthode SuperSAGE : Matsumura et al. (2003) "Gene expression analysis of plant host-pathogen interactions by SuperSAGE" Proc. Natl. Acad. Sci. USA 100, 15718 - 15723 Méthode DeepSAGE : Nielsen et al. (2006) "DeepSAGE -- digital transcriptomics with high sensitivity, simple experimental protocol and multiplexing of samples" Nucleic Acids Res. 34, e133 |
|
|
Méthode MPSS : Brenner et al. (2000) "Gene expression analysis by massively parallel signature sequencing (MPSS) on microbead arrays" Nat.Biotechnol. 18, 630-634 Méthode CAGE : Shiraki et al. (2003) "Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage" Proc Natl Acad Sci U S A 100, 15776 - 15781 Fullwood et al. (2009) "Next-generation DNA sequencing of paired-end tags (PET) for transcriptome and genome analyses" Genome Res. 19, 521 - 532 |
|
|
Méthode RNA-Seq Nagalakshmi et al. (2008) "The Transcriptional Landscape of the Yeast Genome Defined by RNA Sequencing" Science 320, 1344 - 1349 Cloonan et al. (2008) "Stem cell transcriptome profiling via massive-scale mRNA sequencing" Nature Methods 5, 613 - 619 |
|
|
Trapnell et al. (2009) "TopHat: Discovering splice junctions with RNA-Seq" Bioinformatics 25, 1105 - 1111 SOAP: Li et al. (2008) "SOAP: short oligonucleotide alignment program" Bioinformatics 24, 713 - 714 RMAP : Smith et al. (2008) "Using quality scores and longer reads improves accuracy of Solexa read mapping" BMC Bioinformatics 9, 128 MAQ : Li et al. (2008) "Mapping short DNA sequencing reads and calling variants using mapping quality scores" Genome Res. |
|
![]()