
| Les protéines ou régions intrinsèquement désordonnées ("Intrinsically disordered proteins or regions" - IDP/IDR) |
| Tweet |
|
|
1. Présentation générale 2. Plusieurs types de désordre 3. Caractéristiques physico-chimiques des IDP 4. Fonctions biologiques et fonctionnement des IDP 5. Modèles de fonctionnement des IDP 6. Prédicteurs d'IDP et bases de données |
7. "Drug design" et "Drug discovery" : conception de médicaments 8. Les protéines LEA 9. Base de données "LEAPdb" : étude de la relation structure - fonction des protéines LEA (Jaspard E. & Hunault G.) 10. Module bioinformatique : étude des protéines LEA 11. Liens Internet et références bibliographiques |
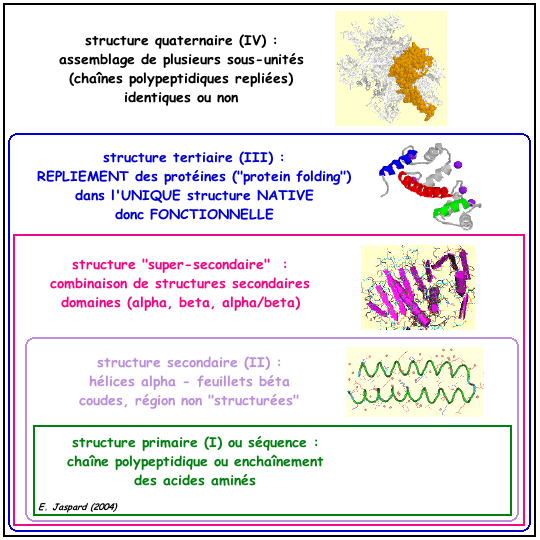
1. Présentation générale Christian ANFINSEN (Prix Nobel 1972) a montré que, dans un environnement approprié : "toute l'information nécessaire au repliement d'une protéine dans sa structure native (donc fonctionnelle) est contenue dans sa séquence primaire (l'enchaînement des acides aminés)". Voir un cours sur le repliement des protéines. Jusqu'au milieu des années 1995, on n'avait donc pas de contre-exemple (reconnu) au dogme : une séquence d'acides aminés => une structure repliée native => une (des) fonction(s) biologique(s) de la protéine. La figure ci-dessous montre les 4 niveaux de structure des protéines.
Acceptation de l'idée de protéines fonctionnelles intrinsèquement désordonnées A partir des années 1980, des résultats expérimentaux montraient que l'absence de structure ou la fléxibilité d'une protéine pouvaient être importantes pour sa fonction biologique (Huber & Bennett, 1983). Mais ces résultats étaient interprétés comme des exceptions, des artéfacts ou bien plus simplement "redécouverts" régulièrement mais décrits avec des dénominations différentes (voir ci-dessous les diverses appellations dans la littérature). Remarque : on voit tout l'intérêt et la force d'une ontologie pour regrouper les différentes terminologies décrivant un seul et même concept. |
Mise en évidence des protéines intrinsèquement désordonnées Au cours des années qui suivirent, l'accumulation d'exemples amenèrent la communauté scientifique à accepter l'idée que des protéines soient fonctionnelles sans avoir une structure native pleinement ordonnée/structurée, aussi bien chez les procaryotes que chez les eucaryotes. Voici 2 exemples :
Ces protéines ou régions intrinsèquement désordonnées ou "intrinsically disordered proteins or regions" - IDP/IDR :
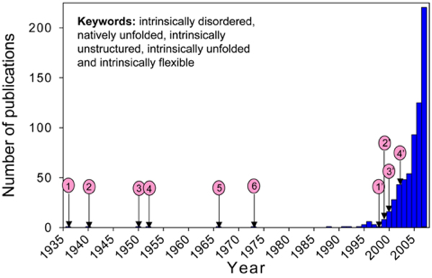
On trouve un grand nombre d'appellations dans la littérature : "natively unfolded proteins", "intrinsically disordered proteins", "intrinsically unstructured proteins", "naturally disordered regions", "intrinsically flexible regions", "rheomorphic proteins", "natively denatured proteins", "mostly unstructured proteins", "natively disordered proteins" mais aussi "pliable", "mobile", "vulnerable", "chameleon", "malleable", "protein clouds", "dancing proteins". Figure ci-dessous : augmentation remarquable du nombre de publications scientifiques dans lesquelles apparaissent des mots synonymes de IDP.
Source : Uversky et al. (2009) Ces appellations ont leur qualité et leur défaut pour rendre compte :
|
Abondance des IDP Afin de comparer l'importance du désordre, via les IDP, chez les Eucaryotes et les Procaryotes, des études bioinformatiques ont été effectuées sur le génome et le protéome de Saccharomyces cerevisiae et Escherichia coli : Les IDP ont été prédites par :
Les résultats ont permis d'obtenir des tendances générales (qui évolueront probablement au fur et à mesure que l'on identifiera de nouvelles IDP) :
L'accumulation d'IDP dans la cellule pourrait induire des problèmes majeurs, par exemple le stress lié à l'accumulation de protéines mal repliées dans le réticulum endoplasmique ("Unfolded protein response" - UPR). L'étude du protéome de l'homme a permis d'établir quelques règles concernant le niveau d'expression des gènes, le temps de demi-vie des ARN messagers, l'adressage / ciblage aux microRNA (miRNA) et l'ubiquitinylation dans le cas des IDP. Ces paramètres jouent un rôle critique dans la disponibilité et la dégradation des IDP dans la cellule. En effet, par rapport aux protéines ordonnées :
Il semble donc que les IDP et leurs transcrits sont présents dans la cellule à une faible concentration et/ou pour un temps court avant d'être marquées pour la dégradation. Cependant, et de manière surprenante, ces 4 paramètres sont inversés pour une grande proportion de protéines fortement désordonnées qui sont fortement exprimées et/ou exprimées de manière constitutive. Ces IDP disposeraient de plus de temps pour agir dans la cellule avant qu'elles ne soient dégradées. En conclusion, bien que les IDP soient capitales pour le fonctionnement cellulaire, des mécanismes protègent la cellule car les IDP sont aussi potentiellement dangereuses puisqu'elles ne sont pas repliées. |
2. Plusieurs types de désordre Par désordre intrinsèque ("intrinsic disorder"), on entend qu'une IDP adopte une multitude de conformations en termes de structures secondaires ou de structures tertiaires. Par opposition, une protéine pleinement structurée/ordonnée a une structure 3D relativement stable, avec des angles de Ramachandran dont les valeurs varient peu autour d'une "position" d'équilibre (avec d'éventuelles variations conformationnelles coopératives). Une IDP ou une IDR est donc inclue dans un ensemble structural dynamique dont les positions des atomes et les angles de Ramachandran du squelette carboné varient de façon significative sans valeur moyenne d'équilibre. Les différentes conformations de cet ensemble subissent des changements non coopératifs. En conclusion, on entend par désordre intrinsèque :
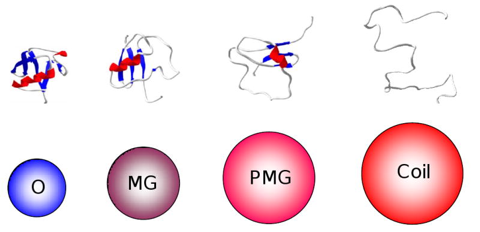
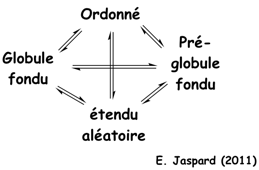
Voir différents modèles du repliement des protéines. Le terme "globule fondu" ("molten globule" - MG) a été proposé par Ohgushi & Wada en 1983 pour décrire un état structural "intermédiaire" du cytochrome C possédant des éléments de structures secondaires semblables à ceux de l'état natif, sans cependant que le coeur hydrophobe de la protéine ait la compacité de l'état natif.
Source : Uversky & Dunker (2010) L'état "globule fondu" est donc un état thermodynamique distinct en équilibre avec les états dénaturés ("unfolded" - D) et natif ("native" - N) dans le chemin du repliement ("protein folding pathway intermediate") : U <=> MG <=> N Figure ci-dessous, le modèle "protein quartet model" (Uversky, 2002) selon lequel la fonction d'une IDP peut émaner de n'importe quelle des 4 conformations ou n'importe quelle des 6 transitions entre ces conformations.
La base de données "DisProt" fournit une sélection d'IDP qui répondent à chacune de ces 10 possibilités. |
a. "Collapsed disorder" : ce type de désordre est représenté par la structure "molten-globule-like", caractérisé par un fort contenu en structures secondaires et une topologie spécifique de ces structures. Ce type de structure est plus sensible aux protéases, possède une grande affinité pour des sondes fluorescentes hydrophobes comme le 8-anilinonaphthalène-1-sulfonate - ANS (ce qui permet de les visualiser) et un grand rayon hydrodynamique (volume bien supérieur aux autres types de structures désordonnées). b. "Partially collapsed disorder" : les IDP "pre-molten-globule-like" possèdent environ 50% de structures secondaires (moins cependant que les IDP "molten-globule-like") et un volume hydrodynamique 3 fois plus important que celui d'une protéine pleinement structurée. Elles lient des sondes hydrophobes fluorescentes (à des taux moins élévés que les IDP "molten-globule-like"). c. "Extended disorder" : ce type de désordre n'a pas de propriété structurale uniforme. Les IDP "random coil-like" ont une très forte charge nette, un volume hydrodynamique élevé (typique de chaînes peptidiques non repliées) et ne possèdent pas de structure secondaire. Les IDP peuvent être caractérisées par de nombreuses méthodes biophysiques et biochimiques, telles que :
Voir un cours sur les méthodes de détermination de la structure des protéines. d. Allostérie et désordre Des modèles (Hilser & Thompson, 2007 - Ferreon et al., 2013) ont montré que le couplage allostérique entre sites de fixation est maximisée quand les domaines qui contiennent un ou plusieurs sites de fixation couplés sont intrinsèquement désordonnés. Ce résultat renforce l'idée d'une prévalence des domaines intrinsèquement désordonnés dans le mode d'action des protéines régulatrices. De plus, il apporte des précisions concernant le schéma classique de la propagation de l'énergie dans les protéines qui prédit que le couplage entre sites serait maximisée lorsqu'un chemin de structures repliées bien définies connecte les sites. Il montre aussi que le mécanisme de couplage conféré par le désordre intrinsèque est indépendant du réseau d'interactions qui relient physiquement les sites de fixation couplés. |
3. Caractéristiques physico-chimiques des IDP Les IDP établissent moins de liaisons intramoléculaires stabilisatrices et sont donc plus dynamiques que les protéines ordonnées. En effet, elles ne possèdent pas suffisamment d'acides aminés non polaires pour former le coeur hydrophobe caractéristique des protéines ordonnées. Cependant, beaucoup d'IDP sont partiellement repliées et ont donc une compacité moyenne (ramenée à la longueur de la chaîne polypeptidique) supérieure à celle d'une chaîne polypeptidique complétement dénaturée / dépliée ("random coil").
Source : Fisher & Stultz (2011)
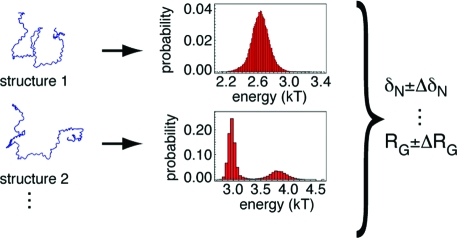
Une IDP ou une IDR est inclue dans un ensemble structural dynamique c'est-à-dire un ensemble plus ou moins vaste de conformations. Cependant, tous les conformères n'ont pas la même probabilité d'existence car ils n'ont pas la même stabilité. Des modèles statistiques Bayésiens tentent de décrire ces différences au sein de tels ensembles.
Source : Fisher et al. (2010) On a recensé envron 460 propriétés physico-chimiques pour les acides aminés. Bon nombre d'entre elles sont "redondantes" ou en tout cas, il existe une forte corrélation entre elles. La charge nette d'une protéine est la propriété physico-chimique qui semble la plus discriminante pour déterminer son degré de désordre et donc s'il s'agit d'une IDP. Cela semble logique puisque plus la charge nette d'une protéine est importante plus les forces de répulsion électrosatiques le sont et plus la chaîne polypeptidique a tendance à être dépliée / désordonnée. Les études statistiques et bioinformatiques de trés grands jeux de données d'IDP (dis XRAY, dis NMR, dis CD, dis Fam32) ont permis de classer les acides aminés du "plus promoteur d'ordre" au "plus promoteur de désordre" : W, F, Y, I, M, L, V, N, C, T, A, G, R, D, H, Q, K, S, E, P |
| Propriétés physico-chimiques les plus discriminantes Source : Dunker et al. (2001) |
Kyte & Doolittle (1982) "Amino acid scale: Hydropathicity" J. Mol. Biol. 157, 105 - 132 Eisenberg et al. (1984) "Amino acid scale: Normalized consensus hydrophobicity scale" J. Mol. Biol. 179, 125 - 142 ------------------------------------------- Bases de données qui recense les échelles de valeurs des propriétés physico-chimiques des acides aminés :
|
| Charge nette | |
| Nombre de contact dans un rayon de 14 Å | |
| Hydropathie - Echelle de Kyte & Doolittle (1982) | |
| Hydropathie - Echelle de Eisenberg et al. (1984) | |
| Flexibilité | |
| Propension à former des feuillets β | |
| Nombre de liaison de coordination | |
| Pourcentage des acides aminés promoteurs de désordre [R + E + S + P] | |
| Encombrement stérique ("bulkiness") | |
| Pourcentage des acides aminés promoteurs d'ordre [C + F + Y + W] | |
| Volume | |
| Réfractivité |
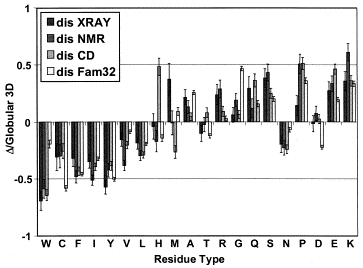
La composition en acides aminés de 4 jeux de données "protéines désordonnées" (dis XRAY, dis NMR, dis CD, dis Fam32) ont été comparés entre eux et avec un jeu de données "protéines ordonnées". La proportion de chaque acide aminé dans chacun des jeux de données a été exprimée par : [nombre de l'acide aminé considéré dans les protéines désordonnées) - (nombre de l'acide aminé considéré dans les protéines ordonnées)] / (nombre de l'acide aminé considéré dans les protéines ordonnées). Dans la figure ci-contre, un pic négatif signifie donc que le jeux de données "protéines désordonnées" considéré contient moins l'acide aminé considéré que le jeu de données "protéines ordonnées".
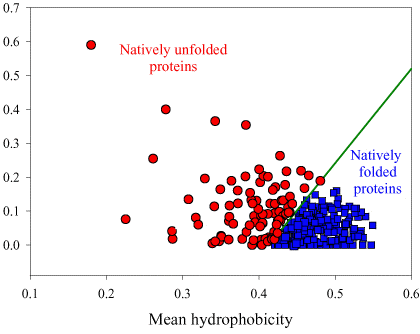
Source : Dunker et al. (2001) Les acides aminés sont rangés en fonction de leur indice de fléxibilité corrigé par le facteur de température ("Debye-Waller factor" ou "B-factor") qui tient compte des mouvements dûs à la châleur sur l'atténuation de la diffraction des rayons X. Celà permet de tenir davantage compte de certains effets de l'environnement sur les acides aminés. L'acide aminé le moins flexible est à gauche (Trp) et le plus flexible est à droite (Lys). Si on représente la valeur absolue de la charge nette moyenne (c'est-à-dire pondérée par la longueur de la chaîne polypeptidique de l'IDP considérée) à pH 7 (<R>) en fonction de la valeur absolue de l'hydrophobicité moyenne (<H>), on obtient un graphique avec deux zones qui correspondent aux IDP et aux protéines structurées, respectivement. Ces zones sont délimitées par une droite d'équation : <H> = [ <R> + 1,151 ] / 2,785 et les IDP sont au dessus de cette ligne.
Source : Uversky et al. (2000) On obtient un graphique équivalent si on représente <R> en fonction de la valeur absolue de l'hydropathie moyenne ("GRand Average of hYdropathy" - <GRAVY>). |
4. Fonctions biologiques et fonctionnement des IDP Les IDP sont impliquées dans des processus de régulation et des voies de signalisation où la fixation à de multiples partenaires jouent un rôle crucial. On les qualifie de "concentrateur" ("hub") en termes de réseaux d'interactions protéine-protéine ("protein–protein interaction (PPI) networks") :
Exemples de "hub" IDP : l'α-synucléine et la protéine p53 Exemples de "hub" structurés : la protéine 14.3.3 et la calmoduline Le désordre structural des IDP confère aux cellules un avantage certain en terme évolutif (Procaryotes vs. Eucaryotes). Exemple : la communication au sein de structures pluricellulaires chez les Eucaryotes. Les IDP sont impliquées dans un grand nombre de fonctions telles que :
Les protéines LEA ("Late Embryogenesis Abundant") sont un exemple typique d'IDP. |
Avantages fonctionnels du désordre Les IDP sont caractérisées par une grande flexibilité et une grande surface accessible au solvant. Ces propriétés sont à la base des fonctions des IDP.
Les IDP ont donc un grand dynamisme structural. Par exemple, les nucléoporines, qui forment le canal des pores nucléaires, possèdent un grand domaine non replié avec des motifs F/G répétés. Au sein du canal, les motifs F/G forment un réseau de polypeptides malléables : certains domaines F/G sont compactés avec une charge nette faible alors que d'autres domaines F/G, structurallement plus dynamiques, sont étendus avec une charge nette élevée. La liaison des IDP à à leurs partenaires induit la transition désordre => ordre qui se traduit par une diminution de la surface accessible (la surface accessible des IDP ne diminue donc pas via des interactions intra-moléculaires mais via l'interaction avec leurs partenaires). Cette transition et l'amplitude de cette diminution dépendent du partenaire lié et augmentent la spécificité de la liaison. Une spécificité accrue de l'interaction entraîne une plus grande réversibilité. C'est un avantage énorme qui explique le rôle des IDP dans les processus de régulation et les voies de signalisation. Voir l'exemple de l'entrée de la colicine ColE9 via les porines OmpF et OmpC chez Escherichia coli. La base de données "DisProt" est un recueil des fonctions des IDP qui sont :
a. S'il n'y a pas d'interaction, on parle de chaînes polypeptidiques entropiques dont la forme est modifiable. Ces IDP peuvent donc relier des domaines fonctionnels d'une même protéine ou de protéines différentes. b. Si l'interaction est permanente, l'IDP se comporte comme :
c. Si l'interaction est transitoire, l'IDP se comporte comme :
|
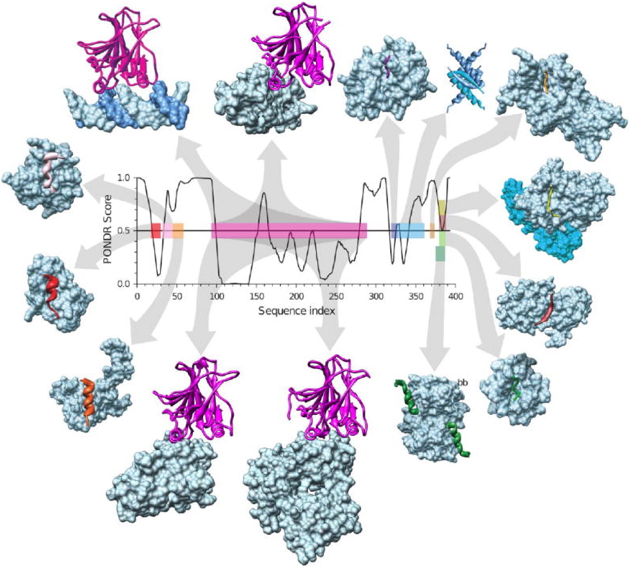
Exemples de "hub" IDP : la protéine p53 p53 est un facteur de transcription d'un grand nombre de gènes codant pour des protéines impliquées dans la protection de la cellule via le cycle cellulaire ou l'apoptose. p53 est constitué de plusieurs domaines (PDB : 1TUP) parmi lesquels :
Au niveau du domaine de transactivation, p53 interagit, entre autres, avec les protéines TFIID, TFIIH, Mdm2, RPA, CBP/p300 et CSN5/Jab1. Au niveau du domaine C-terminal, p53 interagit, entre autres, avec les protéines GSK3β, PARP-1, TAF1, TRRAP, hGcn5, TAF, 14-3-3, S100B(ββ). Voir une liste des molécules avec lesquelles p53 interagit. Figure ci-dessous : quelques-un des partenaires de p53 et localisation des segments d'interaction. Les partenaires à partir de la gauche en haut puis dans le sens des aiguilles sont : l'ADN, 53BP1, gcn5, domaine tet p53, set9, cycline A, sirtuine, domaine bromo-CBP, s100bb, sv40 Large T-antigen, 53BP2, PH, MDM2 et rpa70.
Source : Uversky et al. (2009) Le désordre intrinsèque des régions d'interaction a été prédit par PONDR VLX (voir le paragraphe "Prédicteurs" ci-après).
|
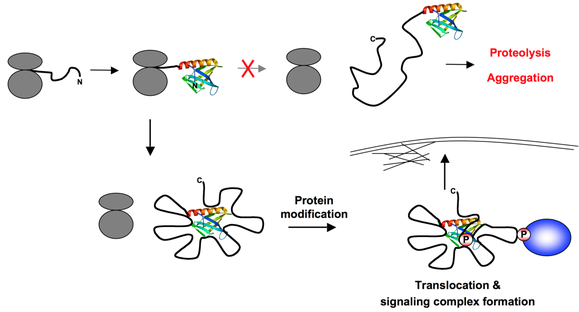
5. Modèles de fonctionnement des IDP Hypothèse de la nucléation par repliement de l'extrémité N-terminale ("N-terminal Folding Nucleation" - NFN) Les protéines qui contiennent un trés grand nombre de sites d'empilement ("large multisite docking proteins" - protéines LMD) comme Gab, p130Cas et les protéines de la famille IRS, facilitent l'assemblage d'énormes complexes protéiques impliqués dans la transduction du signal. Beaucoup de protéines LMD ont un domaine N-terminal structuré ("structured N-terminal domain" - SNTD).
Source : Simister et al. (2011) L'hypothèse NFN propose que, au moment où la chaîne polypeptidique naissante de la protéine LMD (fil noir) émerge du ribosome (en gris), le SNTD se replie rapidement et spontanément. Il sert alors de centre de nucléation pour de nouveaux contacts intramoléculaires spécifiques au sein de la chaîne qui engendrent une forme plus compacte de la protéine. Cette compaction doit probablement préserver la chaîne de la protéolyse et de l'aggrégation. A l'inverse, l'agencement des régions et des boucles empilées génère des régions bien définies de la protéine qui doivent servir de sous-unités fonctionnelles. Des modifications de certaines de ces régions comme la phosphorylation pourrait conduire à la libération de régions empilées, permettant au SNTD de s'engager dans de nouvelles interactions qui permettraient l'ancrage de la protéine LMD dans des compartiments sub-cellulaires specifiques. D'autres modifications sont connues pour engendrer des points d'empilement pour l'interaction avec des partenaires cellulaires, ce qui aboutit probablement à l'assemblage rapide de sous-complexes bien définis sur des boucles spécifiques. Ces diverses propriétés doivent augmenter l'aptitude des cellules à répondre rapidement et sélectivement à différents types de stimuli. |
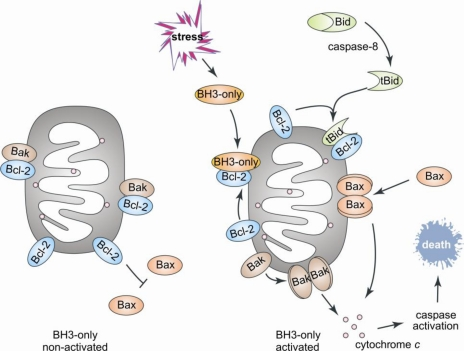
IDR et apoptose Bien que le pourcentage d'identité entre les séquences des protéines de la famille Bcl-2 soit faible, elles possèdent toutes un à quatre domaines conservés appelés domaine BH ("Bcl-2 homology domain"). Les protéines pro-apoptotiques sont sub-divisées en 2 groupes :
Figure ci-dessous : schéma décrivant le rôle des protéines de la famille Bcl-2 dans l'apoptose mitochondriale "intrinsèque" ou activée.
Source : Rautureau et al. (2010) La protéine "BH3-only" Bid établit le lien entre l'apoptose dont la signalisation est sous le contrôle du récepteur de la mort cellulaire ("extrinsèque") et l'apoptose mitochondriale "intrinsèque". Bid est activée par protéolyse par la caspase-8 au niveau de son IDR. |
| FoldIndex | "A simple tool to predict whether a given protein sequence is intrinsically unfolded" | ANCHOR | "Prediction of Protein Binding Regions in Disordered Proteins" | |
| PONDR | "Predictor Of Naturally Disordered Regions" Voir la différence de prédiction entre PONDR VL-XT et PONDR VSL2 (appelé aussi DisProt-VSL2). |
D2P2 | "Database of disordered protein predictions" | |
| SEG | "Prediction of Low Complexity Regions" | MobiDB | "A comprehensive database of intrinsic protein disorder annotations" | |
| IUPRED | "Prediction of Intrinsically Unstructured Proteins" |
IDEAL | "Intrinsically Disordered proteins with Extensive Annotations and Literature" | |
| DISOPRED2 | "The Prediction of Protein Disorder Server" | PrDOS | "Protein DisOrder prediction System" | |
| CSpritz | "Disorder Prediction with CSpritz" | Disembl | "Intrinsic Protein Disorder Prediction" | |
| ESpritz | "Efficient disorder Prediction" |
Autres algorithmes / sites web de prédiction du désordre : GLOBPLOT 2.3, META-Disorder, PONDR FIT ... | ||
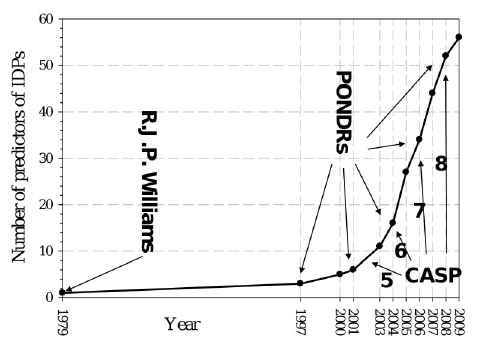
Les IDP possèdent une composition en acides aminés très différente des protéines structurées. Le prédicteur PONDR ("Predictor Of Natural Disordered Regions") s'appuie sur les différentes caractéristiques physico-chimiques des acides aminés (hydropathie, charge nette, hydrophobicité, flexibilité, tendance à former des structures secondaires, ... ) afin d'évaluer le degré de désordre d'une protéine. Voir une description de l'algorithme de PONDR. Le prédicteur IUPRED compare des séquences de régions ordonnées et de régions désordonnées. Il tient compte d'un paramètre supplémentaire discriminant ces deux types de régions : leur capacité à établir des liaisons stables puisque la liaison entre acides aminés tend à diminuer l'énergie interne de la protéine. L'environnement dans lequel se trouve chaque acide aminé est pris en compte : IUPred utilise une fenêtre d'une largeur variable (généralement de 31 acides aminés) qui "glisse" le long de la séquence. Les acides aminés promoteurs de désordre ont une propension moindre du point de vue énergétique à établir des liaisons. L'estimation de ce degré d'énergie se fait en comparant la chaîne polypeptidique testée à un ensemble de protéines globulaires via une matrice d'interactions. En associant l'énergie calculée et la composition en acides aminés environnants, une valeur moyenne de désordre est calculée pour chaque acide aminé. La base de données "DisProt"utilise une fenêtre d'une largeur variable (généralement de 51 acides aminés) qui "glisse" le long de la séquence. Figure ci-dessous : augmentation du nombre de prédicteurs.
Source : Uversky & Dunker (2010) CASP ("Critical Assessment of Techniques for Protein Structure Prediction") est une communauté internationale regroupant plus de 100 laboratoires du monde entier, impliqués dans la prédiction de la structure des protéines. La prédiction des protéines désordonnées a été introduite en 2002 (CASP5). L'expérience d'évaluation critique de la prédiction du désordre intrinsèque des protéines ("The Critical Assessment of protein Intrinsic Disorder prediction" - CAID , le pendant de CASP pour les protéines structurées) a été créé et constitue un "test à l'aveugle" de la communauté scientifique pour établir l'état de l'art dans la prédiction des régions intrinsèquement désordonnées et le sous-ensemble de résidus impliqués dans la fixation.
|
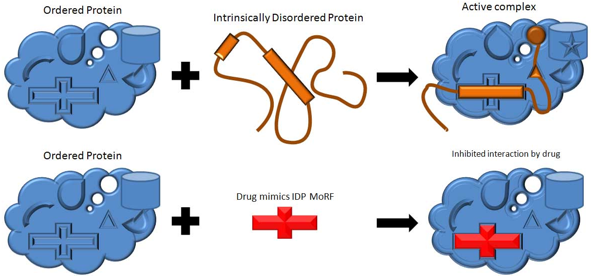
7. "Drug design" et "Drug discovery" : conception de médicaments Les "sites caractéristiques de reconnaissance entre molécules" ("Molecular Recognition Features" - MoRFs) sont de courts peptides d'environ 20 à 30 acides aminés, au sein de longues IDR, qui se fixent spécifiquement à un partenaire cellulaire en subissant une transition désordre ===> ordre. Des molécules mimant ces MoRFs ou leur sites de fixation pourraient être ainsi adressées de façon trés sélective à leurs cibles afin de reproduire et/ou modifier les interactions protéine-protéine. La société "Molecular Kinetics, Inc." a effectué une prédiction de MoRFs sur des IDP et a identifié 35.781 séquences dans le protéome de l'homme susceptibles de déboucher sur des molécules actives. La recherche de MoRFs est un domaine en pleine expansion, notamment en ce qui concerne la prédiction de sites d'IDP qui pourraient être cibles de molécules à visée thérapeutique.
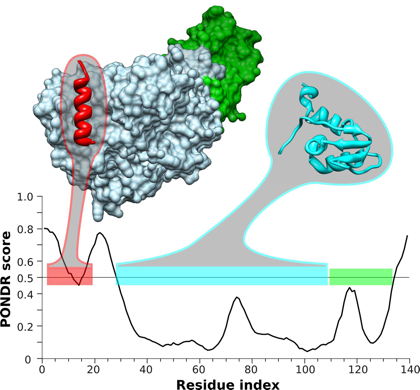
Source : Gorka A. (2011) PONDR VL-XT et ANCHOR sont des algorithmes de prédiction de MoRFs. Figure ci-dessous : comparaison de la structure de la sous-unité β du facteur d'initiation de la traduction aIF2β de Sulfolobus solfataricus prédite par PONDR VSL2 avec la structure déterminée expérimentalement (PDB 2NXU).
Source : Xue et al. (2010) Les scores supérieurs à 0.5 correspondent aux IDR. Les barres de couleur horizontales représentent les régions de structure connue (ou tout du moins probablement structurée) qui sont (du N-terminal vers le C-terminal) :
aIF2β est une IDP avec des régions désordonnées, en particulier l'extrémité N-terminale. Cette région médie la fixation de la sous-unité aIF2γ via une interaction de type MoRF. Il est probable que cette interaction procure aux différents domaines la fléxibilité nécessaire à la reconnaissance entre les molécules. |
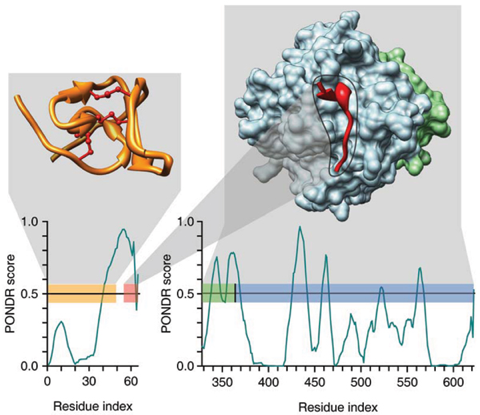
Analyse de l'interaction hirudine - thrombine avec PONDR VL-XT Une région de l'hirudine (PDB 5HIR) impliquée dans l'interaction avec la thrombine (PDB 1NO9) possède un profil particulier et reconnaissable où de courtes régions ordonnées sont entourées d'IDR étendues. Figure ci-dessous à gauche : le domaine N-terminal replié / structuré de l'hirudine (en jaune) avec des ponts disulfure (traits marrons). Une région prédite de type MoRF est représentée par une barre rose.
Source : Dunker et al. (2008) |
| Bases de données de petites molécules et de leur activités biologiques | |
| Pubchem | "A comprehensive repository for small molecules and their biological activities. It includes more than 500,000 bioassay results, 130 million experimental bioassay results, 85 million substances representing 30 million chemically unique compounds." |
| chEMBLdb | "A database that contains chemical properties (e.g. log P, Molecular Weight, Lipinski Parameters) and biological activities (e.g. binding constants, pharmacology and ADMET data) for over 700,000 distinct drug-like small molecules including more than 3 million bioactivity results from the scientific literature." |
| DrugBank | "Displays manually annotated fact sheets of drugs (with chemical, pharmacological and pharmaceutical information) and their targets (with sequence, structure, and pathway) of 6796 drug-like chemicals including 1571 FDA-approved small molecule and biotech (protein/peptide) drugs. Additionally, 4285 non-redundant proteins (i.e. drug targets/enzymes/transporters) are associated with drug entries including 230 richly illustrated drug-action pathways." |
| STITCH : ChemicalProtein Interactions | "A meta-repository to explore known and predicted interactions between more than 300,000 chemicals and their protein targets within the context of protein-protein association networks (2.6 million proteins in 1133 organisms)" |
| GEO: Gene expression omnibus | "A public repository that archives and freely distributes 20,000 microarray-based and sequence-based functional genomics studies including disease-associated experiments." |
| Connectivity Map | "A large collection of genome-wide expression profiles generated with microarrays. It contains expression response of four cancer cell lines to over 1300 bioactive, drug-like molecules. With a simple pattern-matching algorithm available on the web server, drugs can be linked to each other or to diseases and genetic perturbations through signature (dis-)similarity search." |
| ACToR | "An online warehouse that integrates over 500 publicly available chemical toxicity resources including ToxRefDB, in vivo animal toxicity studies and ToxCastDB, 500 high-throughput assays of a thousand chemicals. It contains data on more than 500,000 chemicals and their potential health and environmental risks." |
| SIDER: Side Effects Resource | "A machine-readable side effect resource that connects 888 approved drugs to 1450 recorded adverse effects extracted from public documents and package inserts. Moreover, it integrates and makes available side-effect frequency, and drug and side effect classifications." |
| NPC: NCGC Pharmaceutical Collection | "A comprehensive resource of approved and experimental drugs useful for drug repositioning. In addition to being an electronic resource for computational approaches, it also offers a physical compound collection for high-throughput screening experiments." |
| Source : Iskar et al. (2011) | |
|
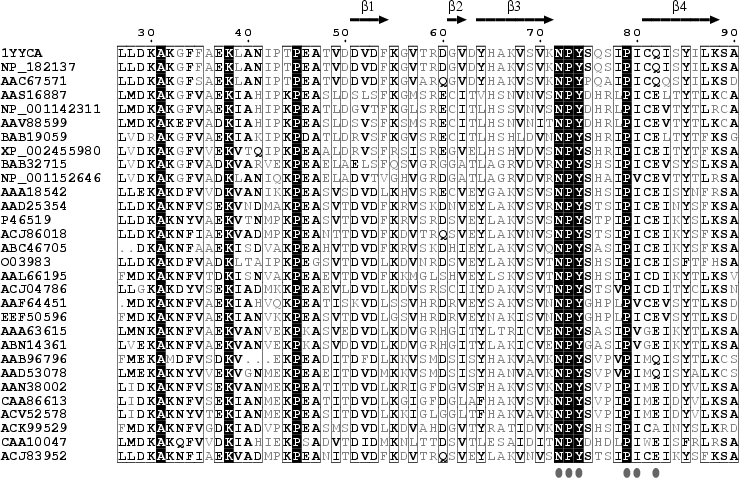
Les protéines LEA ont été mises en évidence dans la phase tardive de l'embryogénèse de semences (d'où leur nom) du coton (Gossypium hirsutum) (Dure, Galau et al.). Leur synthèse et leur accumulation est corrélée à divers types de stress, notamment le stress hydrique (déshydratation, dessication), le stress par le froid et le stress salin. Bien qu'initialement mises en évidence chez les plantes, on en découvre cependant de plus en plus dans d'autres types d'organismes, notamment les organismes anhydrobiotes. Par exemple, des invertebrés comme les nématodes (Aphelenchus avenae), les rotifères, les tardigrades, les chironomides (Polypedilum vanderplanki ) et les arthropodes (Artemia franciscana) qui sont capables de survivre à une déshydratation réversible jusqu'à 2% d'eau dans leurs tissus. Les protéines LEA forment une famille réparties en 12 classes qui correspondent à 8 PFAM. Ces classes se distinguent par l'utilisation des acides aminées et les propriétés physico-chimiques très différentes des protéines qui en découlent. A l'exception de la classe 7, les protéines LEA n'ont pas de structure tri-dimensionnelle établie à l'état natif dans des conditions normales d'hydratation. Les protéines LEA sont donc pour la plupart des IDP. Cette absence de structure tridimensionnelle connue et résolue pose un problème :
|
| Evolution de la classification des protéines LEA | |||||||
| PFAM (name) | Dure et al. (1989) | Bray (1993) | Tunnacliffe & Wise (2007) | Battaglia et al. (2008) | Bies-Esthève et al. (2008) | Hundertmark & Hincha (2008) | LEAPdb Hunault & Jaspard (2010) |
| PF00257 (dehydrin) | D11 | Group 2 | Group 2 | Group 2 | Group 2 | dehydrin | classes 1 to 4 |
| PF04927 (SMPb) | D34 | Group 6 | Group 6 | Group 5A | Group 5 | SMP | class 11 |
| PF03760 (LEA_1) | --- | Group 4 | Group 4 | Group 4A | Group 4 | LEA_1 | class 10 |
| D113 | Group 4B | --- | |||||
| PF03168 (LEA_2) | D95 | --- | --- | Group 5C | Group 7 | LEA_2 | classes 7 & 8 |
| PF03242 (LEA_3) | D73 | --- | Lea5 | Group 5B | Group 6 | LEA_3 | class 9 |
| PF02987 (LEA_4) | D7 | Group 3 | Group 3 | Group 3A | Group 6 | LEA_4 | class 6 |
| D29 | Group 5 | Group 3B | --- | ||||
| PF00477 (LEA_5) | D19 | Group 1 | Group 1 | Group 1 | Group 1 | LEA_5 | class 5 |
| D132 | |||||||
| PF10714 (LEA_6) | --- | --- | --- | Group 6 | Group 8 | PvLEA18 | class 12 |
| PF10714 (ABA-WDS) | --- | --- | --- | Group 7 | --- | --- | --- |
|
|||||||
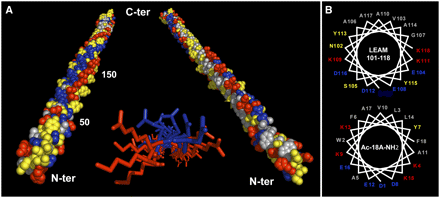
Les protéines LEA et la tolérance à la déshydratation / dessication Leurs fonctions sont encore trés peu connues et encore moins leur mécanisme d'action. Malgré tout, leurs rôles les mieux documentés est l'acquisition d'une tolérance à la dessication par protection des membranes à l'état sec, la séquestration d'ions et la protection de certaines protéines contre la dénaturation. Voir un article qui décrit l'acquisition de la tolérance à la déshydratation de plants de riz transgéniques (transgert du gène HVA1 du blé). Dans une cellule à l'état normal, les protéines LEA n'ont pas de structure tridimensionnelle définie. En revanche, au cours d'un stress comme la déshydratation ("dehydration"), la structure de certaines LEA est enrichie en structures secondaires en particulier en hélices amphiphiles (figure ci-dessous).
Source : Tolleter et al. (2007) Enrichissement en structures secondaires de certaines protéines LEA lors de la dessication Figure ci-dessous, représentations schématiques des structures de la protéine LEA (AaVLEA1) d'Aphelenchus avenae (nématode), obtenues par dynamique moléculaire (champs de force Gromos 96) , pour différents pourcentages en eau (points rouges).
Source : Li & He (2009) Puisqu'aucune structure 3D de LEA n'a été obtenue à ce jour, la structure hypothètique de la protéine AaVLEA1 a d'abord été déterminée par modélisation par homologie ("protein threading - homology modeling") en utilisant comme modèle la structure 3D déterminée pour l'apolipoprotéine AI humaine (PDB : 1AV1). Voir un alignement des séquences de LEA de la classe 7 (E. Jaspard). Un motif conservé au sein de cette classe (comme pour les autres classes) en est la signature. |
| 11. Liens Internet et références bibliographiques |
|
LEAPdb : the database of LEA proteins DisProt : the database of disordered proteins "Institute of Enzymology" - Laboratoire de P. Tompa : un ensemble de présentations Powerpoint des IDP. "Structural and Computational Biology" - European Molecular Biology Laboratory |
|
|
Huber & Bennett (1983) "Functional significance of flexibility in proteins" Biopolymers 22, 261 - 279 Gast et al. (1995) "Prothymosin alpha: a biologically active protein with random coil conformation" Biochemistry 34, 13211 - 13218 Weinreb et al. (1996) "NACP, a protein implicated in Alzheimer's disease and learning, is natively unfolded" Biochemistry 35, 13709 - 13715 |
|
|
Uversky et al. (2009) "Unfoldomics of human diseases: linking protein intrinsic disorder with diseases" BMC Genomics 10, S7 Edwards et al. (2009) "Insights into the regulation of intrinsically disordered proteins in the human proteome by analyzing sequence and gene expression data" Genome Biology 10, R50 Uversky & Dunker (2010) "Understanding protein non-folding" Biochim. Biophys. Acta 1804, 1231 - 1264 |
|
|
Ohgushi & Wada (1983) "'Molten-globule state': a compact form of globular proteins with mobile side-chains" FEBS Lett. 164, 21 - 24 Nakamura et al. (2011) "Thermodynamic and Structural properties of the Acid Molten Globule State of Horse Cytochrome c" Biochemistry 50, 3116 - 3126 |
|
|
Dunker et al. (2001) "Intrinsically disordered protein" J. Mol. Graph. Model 19, 26 - 59 Campen et al. (2008) "TOP-IDP-Scale: A New Amino Acid Scale Measuring Propensity for Intrinsic Disorder" Protein Pept Lett. 15, 956 - 963 Fisher et al. (2010) "Modeling Intrinsically Disordered Proteins with Bayesian Statistics" J. Am. Chem. Soc. 132, 14919 - 14927 Fisher & Stultz (2011) "Constructing ensembles for intrinsically disordered proteins" Curr. Opin. Struct. Biol. 21, 426 - 431 |
|
|
Han et al. (2004) "Evidence for dynamically organized modularity in the yeast protein–protein interaction network" Nature 430, 88 - 93 Simister et al. (2011) "Self-Organization and Regulation of Intrinsically Disordered Proteins with Folded N-Termini" PLoS Biol. 9, e1000591 |
|
|
Uversky V.N. (2002) "Natively unfolded proteins: A point where biology waits for physics" Protein Sci. 11, 739 - 756 Prilusky et al. (2005) "FoldIndex: a simple tool to predict whether a given protein sequence is intrinsically unfolded" Bioinformatics 21, 3435 - 3438 Sickmeier et al. (2006) "DisProt: the Database of Disordered Proteins" Nucleic Acids Res. 35, D786 - D793 Rautureau et al. (2010) "Intrinsically Disordered Proteins in Bcl-2 Regulated Apoptosis" Int. J. Mol. Sci. 11, 1808 - 1824 |
Article |
|
Dunker et al. (2008) "The unfoldomics decade: an update on intrinsically disordered proteins" BMC Genomics 9, S1 Gorka A. (2011) "Prediction and application of intrinsically disordered regions in practice" Jagiellonian University, Pologne Hilser & Thompson (2007) "Intrinsic disorder as a mechanism to optimize allosteric coupling in proteins" PNAS 104, 8311 - 8315 Ferreon et al. (2013) "Modulation of allostery by protein intrinsic disorder" Nature 498, 390–394 |
|
|
Uversky et al. (2000) "Why are "natively unfolded" proteins unstructured under physiologic conditions ?" Proteins 41, 415 - 427 Xue et al. (2010) "Archaic chaos: intrinsically disordered proteins in Archaea" BMC Syst. Biol. 4, S1 Iskar et al. (2011) "Drug discovery in the age of systems biology: the rise of computational approaches for data integration" Curr. Opin. Biotechnol. |
|
|
Protéines LEA Dure et al. (1981) "Developmental biochemistry of cottonseed embryogenesis and germination: changing messenger ribonucleic acid populations as shown by in vitro and in vivo protein synthesis" Biochemistry 20: 4162-4168 Galau & Dure (1981) "Developmental biochemistry of cottonseed embryogenesis and germination: changing messenger ribonucleic acid populations as shown by reciprocal heterologous complementary deoxyribonucleic acid-messenger ribonucleic acid hybridization" Biochemistry 20: 4169-4178 Galau et al. (1986) "Abscisic acid induction of cloned cotton late embryogenesis-abundant (Lea) mRNAs" Plant Mol. Biol. 7: 155-170 Dure et al. (1989) "Common amino acid sequence domains among the LEAP of higher plant" Plant Mol. Biol. 12: 475-486 Galau et al. (1993) "Cotton Lea5 and LEA4 encode atypical late embryogenesis-abundant proteins" Plant Physiol. 101: 695-696 Li D, He X (2009) "Desiccation induced structural alterations in a 66-amino acid fragment of an anhydrobiotic nematode late embryogenesis abundant (LEA) protein" Biomacromolecules 10: 1469-1477. Hunault & Jaspard (2010) "LEAPdb: a database for the late embryogenesis abundant proteins" BMC Genomics 11, 22 Jaspard & Hunault (2012) "Computational and Statistical Analyses of Amino Acid Usage and Physico-Chemical properties of the Twelve Late Embryogenesis Abundant Protein Classes" PLoS One 7, e36968 |
|
|
Necci et al. (2021) "Critical assessment of protein intrinsic disorder prediction" Nat. Methods 18, 472 - 481 |
|
![]()

{kind=link}