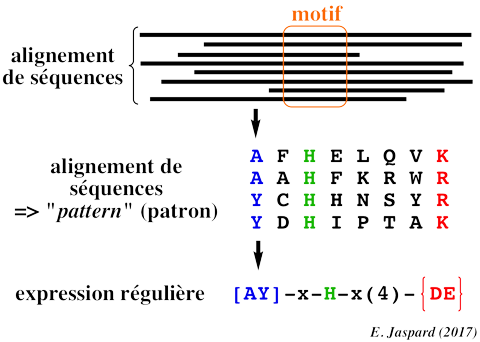
Il existe de nombreux opérateurs ou métacaractères pour les expressions régulières.
En voici quelques exemples.
- L'opérateur "." signifie "n'importe quel caractère" (ici, n'importe quel acide aminé).
- Comme mentionné ci-dessus, le métacaractère "x" remplit la même fonction dans la syntaxe PROSITE.
- L'opérateur "[ ]" signifie "n'importe quel caractère parmi ceux entre les crochets".
- Exemple : [FGKM] n'importe quel acide aminé parmi F, G, K et M.
- L'opérateur "^" est un métacaractère de positionnement ou d'ancrage. Il signifie "commence par". C'est donc la position N-terminale dans le cas d'une séquence d'acides aminés.
- Exemple : ^HHH ou ^H{3} recherche 3 résidus histidine en position N-terminale.
- Exemple : ^[A-DX] signifie "commence par A, B, C, D ou X".
- Remarque : l'opérateur "<" est aussi utilisé pour désigner le premier caractère d'une chaîne de caractères.
- L'opérateur "$" est un métacaractère de positionnement ou d'ancrage. Il signifie "finit par". C'est donc la position C-terminale dans le cas d'une séquence d'acides aminés.
- Exemple: GHS[DE]K$ signifie "finit par la séquence GHS[DE]K".
- Remarque : l'opérateur ">" est aussi utilisé pour désigner le dernier caractère d'une chaîne de caractères.
- L'opérateur "[^]" (^entre crochets) signifie l'exclusion.
- Exemple 1: [^KLY] signifie tous les acides aminés sauf K, L et Y.
- Exemple 2: ^[^G] signifie "qui ne commence pas par G" (expression régulière complémentaire de l'opérateur "^").
- L'opérateur "|" (appelé "pipe") signifie "ou". Exemple : NPN|NPY signifie "contient NPN ou NPY ou les deux"
Les métacaractères "*", "+" , "{ }" et "?" sont des opérateurs de quantification ou quantificateur.
- L'opérateur "*" après un caractère (ou un groupe de caractères) recherche ce caractère zéro fois ou plus.
- L'opérateur "+" signifie au moins 1 fois le caractère (ou le groupe de caractères) qui le précède. Exemple : E+ représente "E" ou "EE" ou "EEE" ou ...
- L'opérateur "{ }" : E{4} = 4 fois E - E{1,3} = 1 à 3 fois E - E{,5} = 0 à 5 fois E - E{7,} = au moins 7 fois E.
- L'opérateur "?" signifie 0 ou 1 fois un caractère (en d'autre terme, l'existence facultative de ce caractère).
- Exemple: CWPN?L$ signifie qu'il peut y avoir ou non le caractère N dans le motif CWP - L (situé en position C-terminale puisqu'il y a l'opérateur $).
Le méta-caractère "\" (back-slash) est utilisé comme caractère d'échappement.
L'opérateur "\w" signifie tout caractère alpha-numérique : équivaut à [_A-Za-z0-9].
L'opérateur "\W" signifie tout caractère autre qu'alphanumérique (c'est le contraire de "\w") : équivaut à [^_A-Za-z0-9].
L'opérateur "\d" signifie que la chaîne de caractères est composé uniquement de chiffre : équivaut à [0-9].
L'opérateur "\D" signifie que la chaîne de caractères n'est pas composé de chiffre (donc composée uniquement de caractères non numériques): équivaut à [^0-9].
Illustration
L'expression régulière ^ATG[ATGC]{30,1000}A{5,10}$ se traduit par :
- Un codon d'initiation ATG au début de la séquence (métacaractère "^" ).
- Suivi de 30 à 1000 ({30,1000}) nucléotides A, T, G ou C (expression régulière [ATGC]).
- Puis 5 à 10 fois le nucléotide A ({5,10}) à la fin de la séquence (métacaractère "$") pour la queue poly-A.
C'est l'expression la plus "simple" pour traduire une séquence d'ARN messager. |