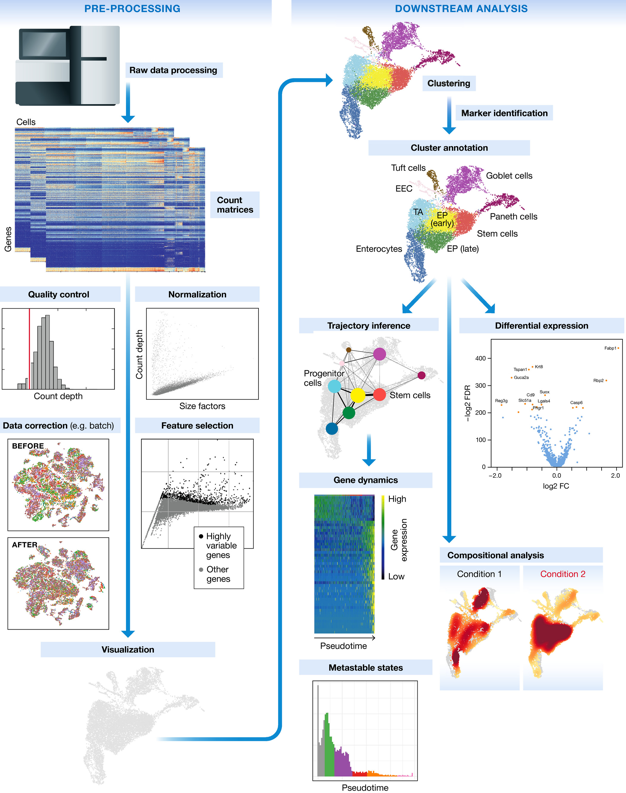
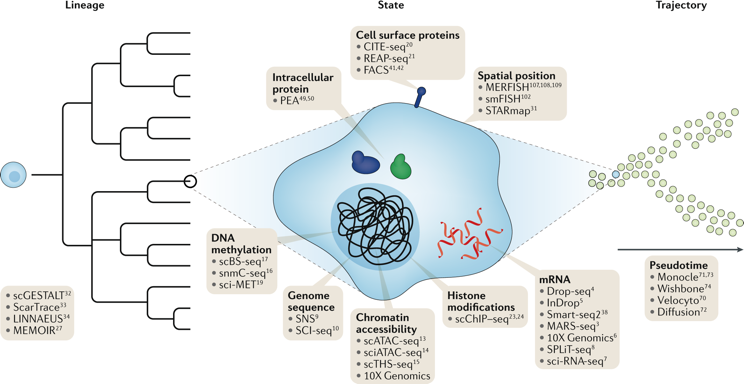
| Quelques méthodes qui caractérisent la lignée cellulaire ("cell lineage") |
- scGESTALT : édition du génome unicellulaire de matrices cibles synthétiques pour le traçage de lignées ("inducible CRISPR-Cas9 system for late barcode editing").
- LINNAEUS : traçage de lignées par édition activée par nucléase de séquences ubiquitaires.
- MEMOIR : mémoire par mutagenèse artificielle avec lecture optique in situ.
|
| Quelques méthodes qui caractérisent l'état actuel d'une cellule ("current state of the cell") |
- Séquençage du génome : SNS, séquençage de noyau unique; SCI-seq, séquençage indexé combinatoire unicellulaire.
- Méthylation de l'ADN : scBS-seq, séquençage au bisulfite unicellulaire; snmC-seq, séquençage de méthylcytosine de noyau unique; sci-MET, indexation combinatoire unicellulaire pour l'analyse de méthylation.
- Accessibilité de la chromatine :
- scATAC-seq ("single cell Assay for Transposase Accessible Chromatin with high-throughput sequencing") ou analyse unicellulaire par séquençage de la chromatine accessible à la transposase.
- sciATAC-seq, analyse par séquençage d'indexation combinatoire unicellulaire de la chromatine accessible à la transposase.
- Modifications de la chromatine : scChIP – seq, immunoprécipitation de la chromatine unicellulaire suivie d'un séquençage.
- ARN messagers : MARS-seq, séquençage monocellulaire d'ARN massivement parallèle; SPLiT-seq, séquençage de transcriptome basé sur la ligature en groupes divisés.
- Protéines intracellulaires : PEA, test d'extension de proximité.
- Protéines de surface : CITE-seq, indexation cellulaire des transcriptomes et des épitopes par séquençage; FACS, tri cellulaire activé par fluorescence ("Fluorescence-activated cell sorting"); REAP-seq, test d'expression d'ARN et de séquençage de protéines.
- Position dans l'espace : MERFISH, hybridation in situ par fluorescence robuste et multiplexée; smFISH, hybridation in situ par fluorescence à molécule individualisée ("Single-molecule Fluorescence in situ Hybridization"); STARmap, cartographie de lecture d'amplicon de transcription résolue spatialement.
|
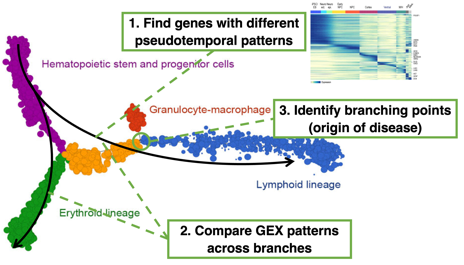
| Quelques méthodes bioinformatiques qui ordonnent les cellules le long d'une trajectoire pseudotemporelle ("trajectory") |
L'inférence de trajectoire ou l'ordre pseudotemporel est utilisée en transcriptomique de cellules individualisées pour (i) décrire la dynamique d'un processus qui se déroule dans les cellules étudiées, puis (ii) pour classer ces cellules en fonction de leur évolution au cours de ce processus.
- Wishbone : algorithme pour aligner les cellules le long des trajectoires de développement avec des branches.
- STREAM : pipeline interactif capable de démêler et de visualiser des trajectoires avec des branchements complexes à partir de données transcriptomiques et épigénomiques de cellules individualisées.
- Velocyto : package pour l'analyse de la dynamique de transcription à partir des données de séquençage d'ARN de cellules uniques. En particulier, il distingue les ARN messagers non épissés et épissés.
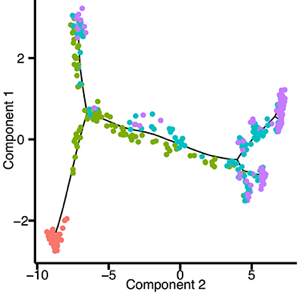
- Monocle : package du langage R qui permet le regroupement, la classification et le comptage des cellules, la construction des trajectoires de cellules uniques et l'analyse d'expression différentielle.
|
| Exemples d'autres méthodes d'analyse de cellules individualisées |
- scCOOL-seq ("Single-Cell chromatin overall omic-scale landscape sequencing") : séquençage de la chromatine de cellules individualisées à l'échelle omique globale.
- scNOMeRe-seq ("Single-Cell nucleosome occupancy, methylome and RNA expression sequencing") : occupation des nucléosomes de cellules individualisées et séquençage du méthylome et des ARN transcrits.
- Analyse simultanée de la modification des histones et de la transcription de cellules individualisées à l'aide de 2 technologies (Paired-Tag & CoTECH).
|
|
10. Liens Internet et références bibliographiques |
Human single-cell
"Human BioMolecular Atlas Program" : programme qui vise à créer un atlas spatial multi-échelles du corps humain à une résolution unicellulaire en diffusant des ressources à la communauté.
Collection d'articles du journal Nature décrivant des travaux issus du HuBMAP.
Human Cell Atlas
Human Protein Atlas : The single cell type section
Data Coordination Platform of the Human Cell Atlas |
HuBMAP
Collection
HCA
HPA
Aller au site |
Manuel de réference "Single-cell best practices"
Single Cell Portal
"A Roadmap for Cyber-Secure Biological Systems" |
Manuel
SCP
Aller au site |
Recueil GitHub de dizaines d'algorithmes d'estimation du pseudo-temps de cellules individualisées
Programme scbean : modèles pour l'analyse d'ensembles de données unicellulaires à grande échelle (notamment réduction de dimensionnalité, suppression des effets de lot et transfert d'étiquettes de type cellulaire).
UMAP : illustration de cette technique de réduction de dimensionnalité.
CytoSPACE : High Resolution Alignment of Single-Cell and Spatial Transcriptomes |
Aller au site
scbean
UMAP
CytoSPACE |
"Introduction to lab-on-a-chip"
10xGenomics : différents algorithmes d'analyse des données "single cell"
Suite logicielle d'analyse d'échantillons uniques "Smart-seq2" :
- Conçu par la plateforme de coordination des données de l'Atlas des cellules humaines ("Data Coordination Platform of the Human Cell Atlas") pour traiter les données de séquençage des ARN de cellules individualisées.
- "Smart-seq2" est écrit en langage de script WDL ("Website Description Language").
|
Aller au site
10xGenomics
Smart-seq2 |
Ensemble 1 de ressources sur la microfluidique
Ensemble 2 de ressources sur la microfluidique |
Ensemble 1
Ensemble 2 |
O'Kane & Gehring (1987) "Detection in situ of genomic regulatory elements in Drosophila" Proc. Natl Acad. Sci. USA 84, 9123 - 9127
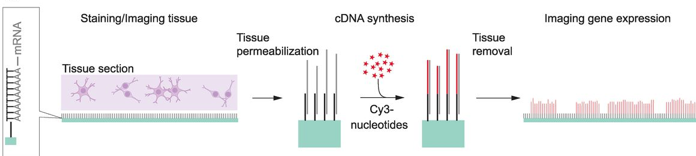
Cet article est l'un des tous premiers à décrire la transcription des gènes dans l'espace ("spatial transcriptomics"). |
Article |
|
1ère publication de la technique "Single-cell"
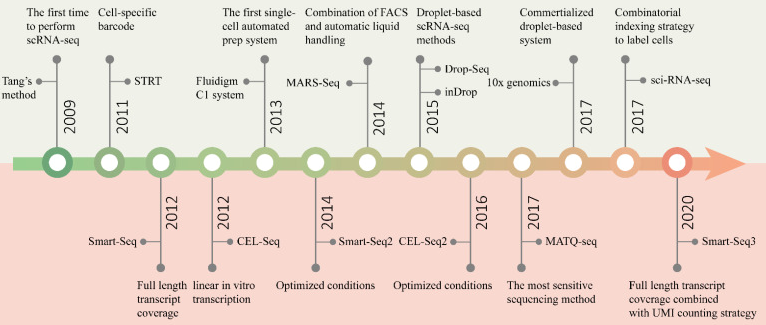
Tang et al. (2009) "mRNA-Seq whole-transcriptome analysis of a single cell" Nat. Methods. 6, 377 - 382 |
Article |
|
Boja et al. (2014) "Integration of omics sciences to advance biology and medicine" Clin. Proteomics 11, 45
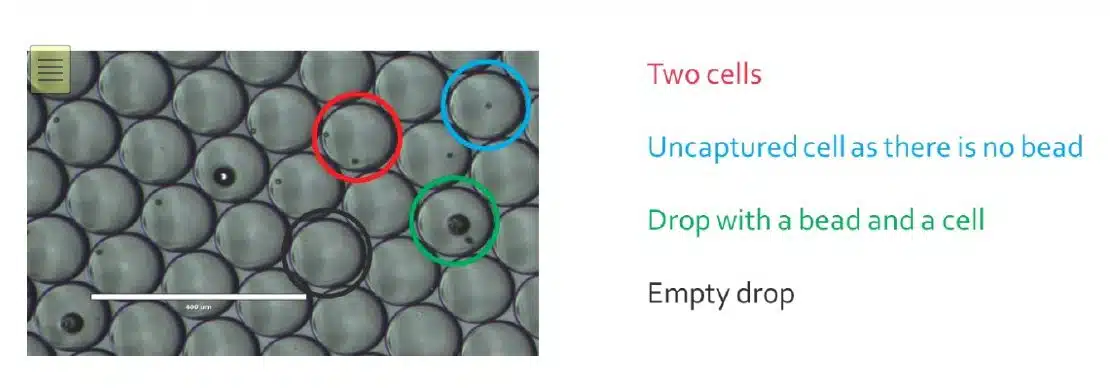
Klein et al. (2015) "Droplet barcoding for Single-Cell transcriptomics applied to embryonic stem cells" Cell 161, 1187 - 1201 |
Article
Article |
|
Van Emon J.M. (2016) "The Omics Revolution in Agricultural Research" J. Agric. Food. Chem. 13, 36 - 44
Reuter et al. (2016) "Simul-seq: combined DNA and RNA sequencing for whole-genome and transcriptome profiling" Nat. Methods 13, 953 - 958
Stahl et al. (2016) "Visualization and analysis of gene expression in tissue sections by spatial transcriptomics" Science 353, 78 - 82 |
Article
Article
Article |
|
Prakadan et al. (2017) "Scaling by shrinking: empowering Single-Cell 'omics' with microfluidic devices" Nat. Rev. Genet. 18, 345 - 361
Haque et al. (2017) "A practical guide to Single-Cell RNA-sequencing for biomedical research and clinical applications" Genome Med. 9, 75
Smith et al. (2017) "UMI-tools: modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy" Genome Res. 27, 491 - 499 |
Article
Article
Article |
|
Noor et al. (2019) "Biological insights through omics data integration" Curr. Opin. Sys. Biol. 15, 39 - 47
Stuart & Satija (2019) "Integrative Single-Cell analysis" Nat. Rev. Genet. 20, 257 - 272
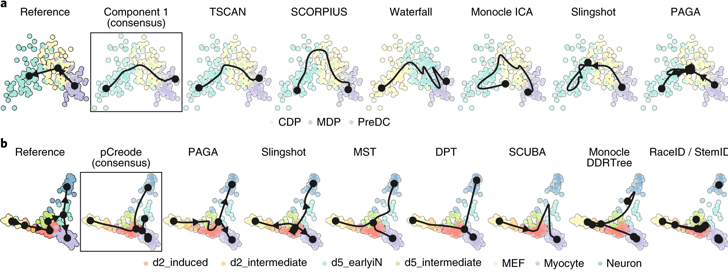
Saelens et al. (2019) "A comparison of Single-Cell trajectory inference methods" Nat. Biotech. 37, 547 - 554
Luecken et al. (2019) "Current best practices in Single-Cell RNA-seq analysis: a tutorial" Mol. Syst. Biol. 15, e8746 |
Article
Article
Article
Article |
Wilbrey-Clark et al. (2020) "Cell Atlas technologies and insights into tissue architecture" Biochem. J. 477, 1427 - 1442
Bond et al. (2021) "Molecular omics resources should require sex annotation: a call for action" Nat. Methods 18, 585 - 588 |
Article
Article |
|
Moses & Pachter (2022) "Museum of spatial transcriptomics" Nat. Methods 19,534 -546
Pan et al. (2022) "Microfluidics Facilitates the Development of Single-Cell RNA Sequencing" Biosensors 12, 450
Yoon & Lee (2022) "Integration of Genomic Profiling and Organoid Development in Precision Oncology" Int. J. Mol. Sci. 23, 216
Jovic et al. (2022) "Single-cell RNA sequencing technologies and applications: A brief overview" Clin. Transl. Med. 12, e694 |
Article
Article
Article
Article |
|
Ahlmann-Eltze & Huber (2023) "Comparison of transformations for Single-Cell RNA-seq data" Nat. Methods 20, 665 - 672
Heumos et al. (2023) "Best practices for single-cell analysis across modalities" Nat. Rev. Genet. 24, 550 - 572
Yang et al. (2023) "Rapid droplet-based mixing for single-molecule spectroscopy" Nat. Methods 20, 1479 - 1482 |
Article
Article
Article |
|
Tarhan et al. (2023) "Single Cell Portal: an interactive home for Single-Cell genomics data" bioRxiv - preprint
Wang et al. (2023) "The Evolution of Single-Cell RNA Sequencing Technology and Application: Progress and Perspectives" Int. J. Mol. Sci. 24, 2943
Georges et al. (2023) "Comparing the transcriptome of developing native and iPSC-derived mouse retinae by single cell RNA sequencing" Scientific Reports 13
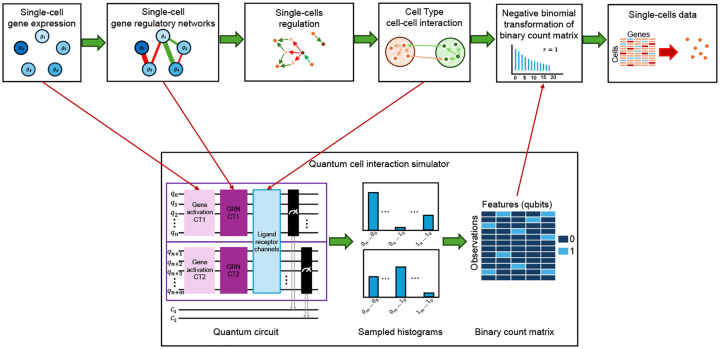
Roman-Vicharra & Cai (2023) "Quantum gene regulatory networks" npj Quantum Information 9, 67 |
Article
Article
Article
Article |
|
Analyses multi-omiques basées sur les cellules individualisées ("Single-Cell multi-omics")
Vandereyken et al. (2023) "Methods and applications for Single-Cell and spatial multi-omics" Nat. Rev. Genet. 24, 494 - 515
Baysoy et al. (2023) "The technological landscape and applications of Single-Cell multi-omics" Nat. Rev. Mol. Cell Biol. 24, 695 - 713
Deng et al. (2023) "Microtechnologies for Single-Cell and spatial multi-omics" Nat. Rev. Bioeng. 1, 769 - 784
Yue et al. (2023) "A guidebook of spatial transcriptomic technologies, data resources and analysis approaches" CSBJ 21, 940 - 955
Lim et al. (2024) "Advances in single-cell omics and multiomics for high-resolution molecular profiling" Exp. Molec. Med.
Cui et al. (2024) "scGPT: toward building a foundation model for single-cell multi-omics using generative AI" Nat. Methods 21, 1470 - 1480
You et al. (2024) "Systematic comparison of sequencing-based spatial transcriptomic methods" Nat. Methods 21, 1743 - 1754
Wu et al. (2024) "Simultaneous single-cell three-dimensional genome and gene expression profiling uncovers dynamic enhancer connectivity underlying olfactory receptor choice" Nat. Methods 21, 974 - 982
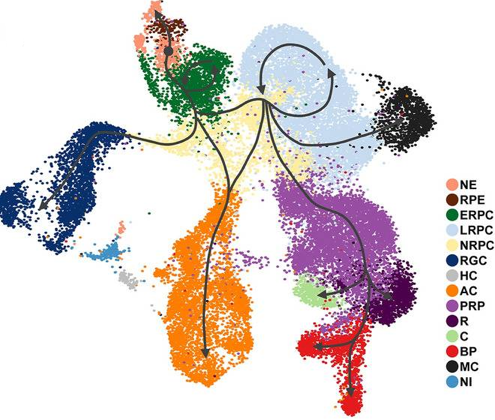
Cheng et al. (2025) "PHLOWER leverages single-cell multimodal data to infer complex, multi-branching cell differentiation trajectories" Nat. Methods 22, 2328 - 2336 |
Article
Article
Article
Article
Article
Article
Article
Article
Article |
Sittipongpittaya et al. (2025) "Protein sequencing with single amino acid resolution discerns peptides that discriminate tropomyosin proteoforms" J. Proteome Res. 24
Joly et al. (2025) "Large-scale single-molecule analysis of tau proteoforms" biorXiv
Marx V. (2025) "Is single-molecule protein sequencing here yet ?" Nature Methods 22, 1623 - 1628
Yazicigil et al. (2025) "Improving engineered biological systems with electronics and microfluidics" Nat. Biotechnol. 43, 1067 - 1083
Manetsch et al. (2025) "A tweezer array with 6,100 highly coherent atomic qubits" Nature 647, 60 - 67 |
Article
Article
Article
Article
Article |
Hevdeli et al. (2026) "CellMentor: cell-type aware dimensionality reduction for single-cell RNA-sequencing data" Nat. Commun. 17, 396
Palastea et al. (2026) "AI-based prediction of gene expression in single-cell and multiscale genomics and transcriptomics" Int. J. Mol. Sci. 27, 801
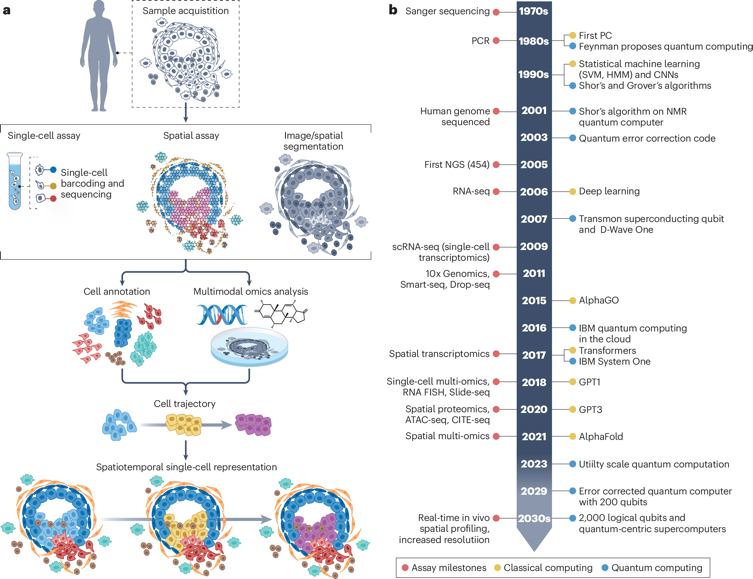
Bose et al. (2026) "Advancing single-cell omics and cell-based therapeutics with quantum computing" Nat. Rev. Mol. Cell Biol. doi: 10.1038/s41580-025-00918-0 |
Article
Article
Article |

{kind=link}