1. Les domaines en omique
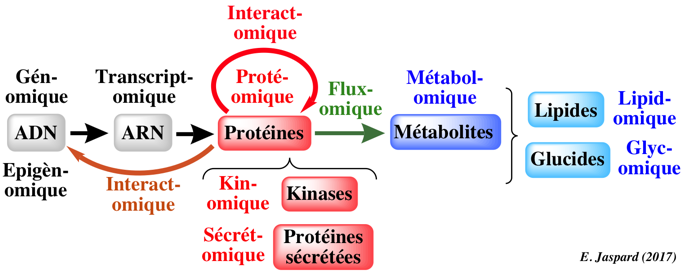
Il existe de nombreux sous-domaines scientifiques biologiques dont le nom a été créé avec le suffixe "omique".
En voici les principaux exemples : génomique - métagénomique - épigénomique - transcriptomique - épitranscriptomique - translatomique - protéomique - métabolomique - interactomique - connectomique - fluxomique - integromique - glycomique - glycoprotéomique - lipidomique - pharmacogénomique, ...
- Les anglo-saxons emploient le suffixe "omics".
- Voir : "List of omics topics in biology".
- La protéogénomique est l'analyse intégrative des données de génomique, de transcriptomique, de protéomique et de modifications post-traductionnelles.
Les figures ci-dessous montrent la répartition des publications (articles) scientifiques dans la base de données bibliographique PubMed qui mentionnent un ou plusieurs domaine(s) en "omique" :
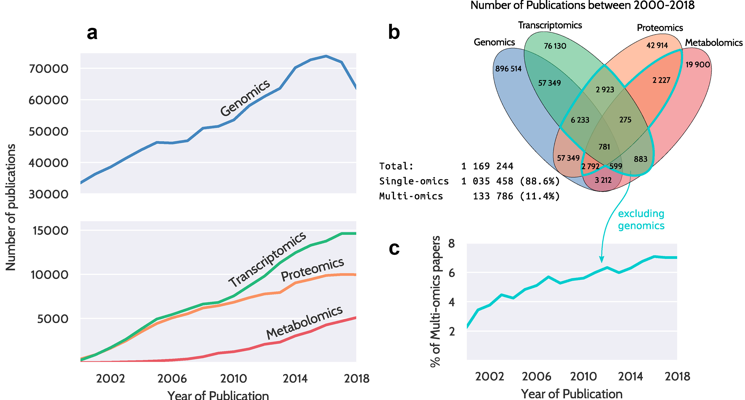
Source : Noor et al. (2019)
- (a) Nombre total d'articles par année depuis 2000 dans 4 domaines en "omique".
- (b) Diagramme de Venn montrant le chevauchement des articles qui mentionnent un ou plusieurs domaine(s) en "omique". Les approches "multiomiques" les plus courantes sont pour l'instant [génomique + protéomique] et [génomique + transcriptomique] qui représentent plus de 10% des articles.
- (c) Pourcentage de publications "multiomiques" mentionnant au moins deux des trois domaines transcriptomique, protéomique et métabolomique (la génomique est omise).
Tous ces domaines évoluent très rapidement. Ils sont de plus en plus intégrés : on peut émettre l'hypothèse qu'ils se fondront en une discipline générale peut-être gérée par une intelligence artificielle.