| Quelques méthodes d'études des interactions protéine-protéine ("Protein-Protein Interaction" - PPI) |
1. La relation structure - fonction des macromolécules biologiques 2. Quantification de l'interaction entre macromolécules biologiques (KA et KD) 3. Notions élémentaires de protéomique a. Caractéristiques générales des protéomes 4. Caractéristiques des méthodes pour détecter et prouver les PPI a. Comparaison de diverses méthodes pour l'étude des PPI 5. Quelques méthodes de pointe pour l'analyse des PPI a. La méthode ascorbate peroxidase (APEX) |
d. La résonance plasmonique de surface (SPR) 6. Titrage calorimétrique isotherme (ITC) a. Principe de l'ITC 7. La photométrie de masse ("Mass photometry") a. Principe 8. Notion de proximité induite chimiquement 9. La prédiction des interactions protéine-protéine par apprentissage profond 10. Liens Internet et références bibliographiques |
1. La relation structure - fonction des macromolécules biologiques a. Les différents types de ligands La molécule qui se fixe sur une autre macromolécule biologique est appelée ligand de manière générique. Un ligand peut être n'importe quel type de molécule biologique :
|
b. Propriétés physico-chimiques des macromolécules biologiques qui modulent et contrôlent leurs interactions Les interactions physiques entre les molécules d'une cellule ou d'un compartiment sub-cellulaire traduisent, entre autre, l'aptitude structurale de ces molécules à se reconnaître.
Exemples :
|
c. Forces de liaison qui maintiennent la structure des macromolécules Ces forces sont non covalentes ou covalentes (ponts disulfures dans le cas des protéines) et très variées en nombre et du point de vue énergétique. Elles sont intimement liées aux propriétés physico-chimiques des résidus d'acides aminés donc liées aux conditions cellulaires (pH, température, viscosité, pression). Tous ces paramètres sont maintenus relativement constants dans la cellule.
Ces paramètres physico-chimiques contrôlent ces équilibres, donc la flexibilité ou dynamique conformationnelle de toutes les molécules biologiques.
|
d. La notion d'affinité entre macromolécules biologiques C'est la caractéristique qui traduit la propension, dans un environnement et des conditions cellulaires donnés, de 2 (ou plus) macromolécules biologiques à se reconnaître et à interagir de manière réversible. Outre la complémentarité de structure, le paramètre clé de l'interaction entre molécules est leur concentration respective.
L'affinité de liaison est influencée par les paramètres physico-chimiques qui influencent la structure des macromolécules biologiques qui interagissent :
|
2. Quantification de l'interaction entre macromolécules biologiques (KA et KD) Toute réaction d'association (inversement de dissociation) entre 2 (ou plus) molécules M1 et M2 peut s'écrire : M1 + M2 <=> M1-M2 Cette réaction d'association est régie par une constante d'association KA (inversement de dissociation KD) quantifiable si on dispose d'une méthode ou d'une technique permettant :
L'équilibre de fixation d'un ligand L sur une protéine P correspond à la réaction : Vitesse d'association : va = ka . [P].[L] - Vitesse de dissociation : vd = kd . [PL]
A l'équilibre, les vitesses sont égales :
Source : Xing et al. (2016) Voir un développement important : équilibre de fixation d'un ligand sur une protéine et représentation de Scatchard. |
3. Notions élémentaires de protéomique a. Caractéristiques générales des protéomes La protéomique a pour but d'identifier (et de quantifier) l'ensemble des protéines synthétisées ou protéome, à un moment donné et dans des conditions données au sein d'un tissu, d'une cellule ou d'un compartiment cellulaire. Le protéome est extrêmement complexe à plusieurs titres :
|
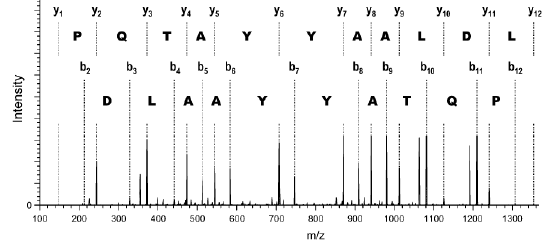
b. Principe de la spectrométrie de masse L'ionisation électronique (souvent appelée impact électronique) et l'ionisation chimique sont les principales méthodes d'ionisation. Dans le cas de l'ionisation électronique, l'échantillon est introduit dans une enceinte sous vide, il y est vaporisé puis soumis au bombardement d'un canon à électrons de grande énergie. Un électron est arraché aux molécules et on obtient une espèce qui est à la fois un cation (ion positif) et un radical libre (nombre impair d'électrons), que l'on appelle ion moléculaire M+. : M + e- (énergie 70 eV) <=> M+. + 2 e- L'énergie du faisceau ionisant fragmente l'ion moléculaire par rupture des liaisons les plus faibles avant les liaisons les plus fortes et donne naissance à des ions positifs de masses plus faibles, qui pourront être fragmentés à nouveau (exemple : spectrométrie de masse dite en tandem - MS/MS). Ces ions sont ensuite accélérés dans un champ électrique et/ou magnétique, puis dirigés entre les pôles d'un aimant selon une trajectoire circulaire qui dépend de leur rapport masse/charge [m/z]. En faisant varier le champ électrique, on fait varier la vitesse des ions moléculaires et on peut les faire ainsi parvenir au détecteur par ordre croissant de rapport [m/z]. Le tri des ions s'effectue :
On obtient un grand nombre de pics, tous de masse inférieure à celle de l'ion moléculaire. Cet ensemble constitue un diagramme de fragmentation. Les groupements fonctionnels possèdent un diagramme de fragmentation qui leur sont propres.
Dans un spectre de masse, la hauteur relative des pics indique l'abondance relative des espèces. |
Voir un développement de la protéomique. Voir des travaux dirigés en ligne : "Applications et résultats de la protéomique : exemple de la RuBisCO" |
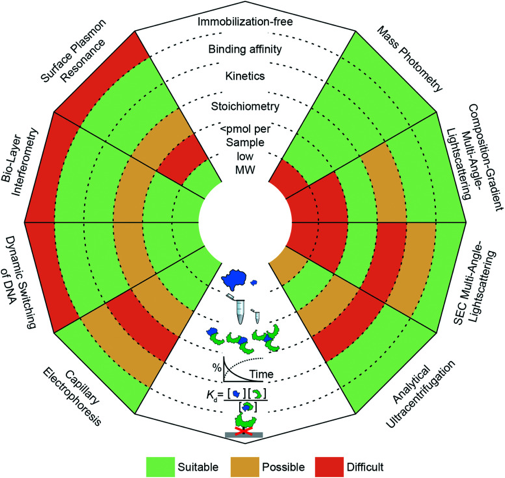
4. Caractéristiques des méthodes pour détecter et prouver les interactions protéine-protéine a. Comparaison de diverses méthodes pour l'étude des interactions protéine-protéine La figure suivanre compare des méthodes optiques dites "sans marquage" : SPR "Surface Plasmon Resonance", BLI ("Biolayer Interferometry"), commutation dynamique des couches d'ADN ("dynamic switching of DNA layers"), CE, AUC, SEC-MALS, CG-MALS et photométrie de masse.
Source : Soltermann et al. (2021) CG-MALS : "Composition-gradient multi-angle light scattering"; AUC : "Analytical ultracentrifugation" |
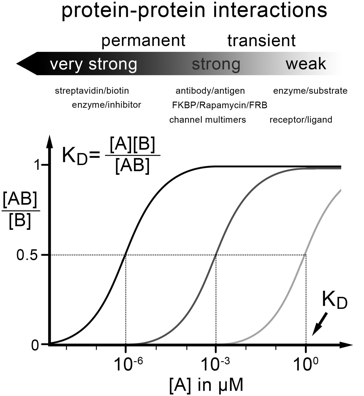
b. Critères de choix d'une technique pour mesurer les interactions protéine-protéine Les techniques sans marquage ou sans immobilisation sont employées autant que possible car :
L'aptitude d'une technique à étudier une large gamme de concentrations permet de mesurer une large gamme de valeurs de KD.
|
Les techniques qui mesurent la cinétique d'atteinte d'un équilibre de fixation entre molécules permettent de déterminer la valeur de KD et celles des constantes de vitesse d'association (ka) et de dissociation (kd). Ces techniques impliquent de quantifier la concentration des molécules non liées et liées (complexe) à différents temps jusqu'à atteindre l'équilibre. Il existe 2 procédures :
Les valeurs de ka et kd peuvent s'échelonner de quelques millisecondes à des heures : le suivi de la cinétique de réactions avec une sensibilité élevée sur de longues périodes exige une correction permanente des lignes de bases et du bruit expérimental. Les derniers critères de choix, et non les moindres, sont :
|
c. Aperçu des méthodes biologiques (complémentation) & biochimiques
Techniques d'immunoprécipitation
|
d. Aperçu des méthodes physiques
Cette interaction hautement spécifique permet d'isoler les protéines marquées par Strep en une étape à partir de lysats cellulaires bruts. De plus, les conditions d'élution de Strep-tag sont "douces" : ce marquage permet d'isoler des protéines fonctionnelles.
|
e. Méthodes bioinformatiques
|
|
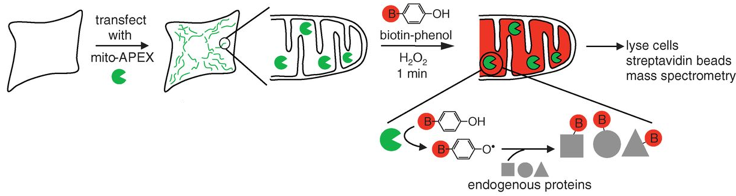
5. Quelques méthodes de pointe pour analyser les interactions protéine-protéine a. La méthode ascorbate peroxidase (APEX) La plupart des protéines sont localisées au sein d'organites entourés de membrane(s). Cette localisation subcellulaire des protéines est un élément clé de leur spécificité de fonction(s). Une méthode de marquage par la biotine, basée sur l'ascorbate peroxidase (APEX) modifiée par ingénierie ("engineered ascorbate peroxidase (APEX)-based proteins proximity labeling") permet de localiser et d'identifier des protéines situées à proximité les unes des autres dans la cellule, et donc d'identifier leurs interactions. L'ascorbate peroxidase (APX1 - E.C. 1.11.1.11) est une oxydoréductase qui catalyse la réaction : L-ascorbate + H2O2 <=> déshydro-ascorbate + 2 H2O L'APEX est un monomère d'environ 28 kDa sans pont disulfure ni site de fixation du calcium. |
α. Principe de la méthode L'APEX est active dans tous les compartiments cellulaires.
Cette réaction permet de biotinyler les protéines adjacentes (dans une sphère de rayon ≈ 20 nm) à l'APEX exprimée localement.
Les protéines sont ensuite éluées, séparées sur gel et identifiées par spectromètrie de masse.
Source : Rhee et al. (2013) - B = biotine Remarque : l'APEX a une sensibilité limitée qui exclut les applications nécessitant un faible taux d'APEX : pour cette raison, une enzyme appelée APEX2 a été développée par ingéniérie et elle a une activité plus importante dans les cellules. |
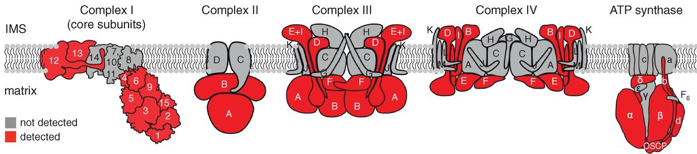
β. Illustration de l'identification d'interactions entre protéines avec l'APEX Une étude a utilisé un marquage spécifique des protéines de la matrice de la mitochondrie par la biotine dans des cellules vivantes : l'ensemble des membranes et des complexes protéiques étaient donc intacts et ainsi les relations spatiales (les interactions) entre les protéines ont été préservées.
Cette étude a permis :
Source : Rhee et al. (2013) |
b. La fluorescence par complémentation bimoléculaire (BiFC) Une protéine de fusion est une combinaison de différentes protéines ou régions de protéines. Elle est synthétisée par une construction d'ADN contenant les cadres de lecture ouverts ("Open Reading Frame" ou ORF) codant les protéines ou régions de protéines concernées.
α. Principe de la méthode La fluorescence par complémentation bimoléculaire ("Bimolecular fluorescence complementation" - BiFC) permet de visualiser une interaction protéine-protéine dans les cellules sans traitement particulier de ces cellules.
|
β. Illustration
Source : Kodama & Hu (2012)
Voir un vecteur d'expression pour l'obtention d'une protéine de fusion avec le fragment C-terminal de la protéine fluorescente jaune Venus. Diverses variantes de la méthode BiFC ont été développées :
|
c. Alternatives au marquage fluorescent par de grosses protéines de fusion Diverses stratégies sont développées pour marquer les protéines d'intérêt ("Protein Of Interest" - POI) dans les cellules vivantes. Elles ont pour but d'accroître l'efficacité du marquage, de minimiser (voire abolir) l'incidence de la taille de la molécule fluorescente ajoutée et, dans certains cas, coder génétiquement l'incorporation d'acides aminés non naturel réactifs dans la séquence d'une protéine d'intérêt. |
α. Fixation de petits colorants organiques fluorescents
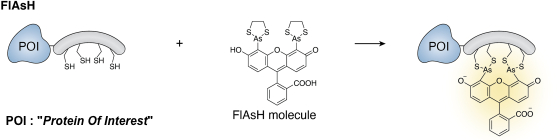
Exemple : le petit ligand FlAsH ("Fluoroscein Arsenical Hairpin", < 700 Da) composé d'une molécule de fluorescéine et de 2 atomes d'arsenic (As).
Source : de Luis et al. (2025)
|
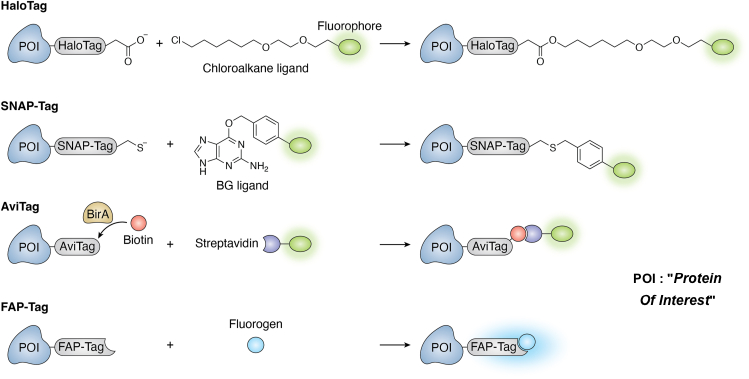
β. Etiquettes peptidiques pour remplacer les protéines fluorescentes volumineuses De manière équivalente aux protéines fluorescentes de fusion codées génétiquement, ces étiquettes nécessitent la biosynthèse de chaînes polypeptidiques de fusion qui, du fait de leur taille, peuvent altérer la fonction de la protéine d'intérêt. Ces étiquettes peptidiques sont utilisées dans des applications FRET. Étiquette Halo
Étiquette SNAP
Étiquette CLIP
Source : de Luis et al. (2025) Étiquette Avi
Étiquette FAP
|
γ. Acides aminés non naturels, chimie "click" et chimie "bio-orthogonale"
Leur réactivité extrêmement élevée et leur compatibilité avec les cellules vivantes rend ces méthodes idéales pour le marquage rapide, spécifique et efficace des molécules biologiques. Pour accroître ces avantages dans les cellules, on incorpore aux protéines d'intérêt des AANN "cliquables" portant des groupements réactifs dans leurs chaînes latérales. Exemples de réactions qui ajoutent des groupements réactifs aux AANN "cliquables" :
|
δ. Codage génétique de l'incorporation d'AANN réactifs par expansion du code génétique Le développement par ingénierie des protéines de paires [ARNt / aminoacyl-ARNt synthétase modifiée] orthogonales et la modification de l'expression d'un codon STOP dirigent l'incorporation co-traductionnelle et site-spécifique d'AANN réactifs dans la séquence d'une protéine d'intérêt. Voir des exemples de paires [ARNt / aminoacyl-ARNt synthétase modifiée] orthogonales. Exemple de l'incorporation de la pyrrolysine Les systèmes qui s'appuient sur la modification de l'expression du codon STOP ambre (UAG) permettent d'incorporer une pyrrolysine grâce :
La pyrrolysine (Pyl) est donc intégrée dans une séquence peptidique : ARNtPyl + L-pyrrolysine + ATP -> L-pyrrolysyl-ARNtPyl + AMP + diphosphate
|
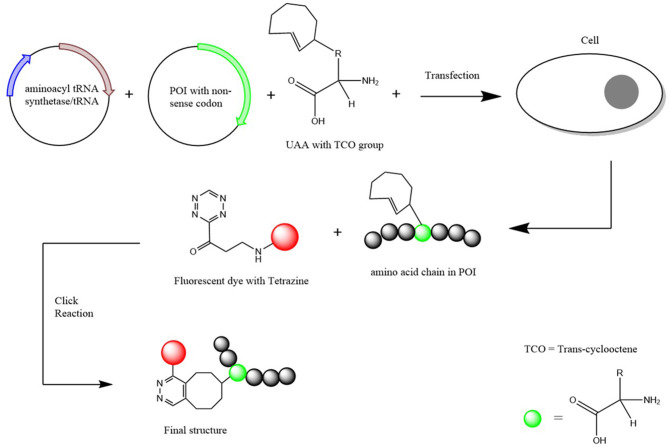
ε. Illustration de ces systèmes Des cellules sont transfectées avec :
L'AANN réactif est ainsi incorporé dans la séquence de la protéine d'intérêt.
Source : Laxman et al. (2021) Puis une petite molécule fluorescente (portant un groupe tétrazine dans l'exemple) est ajouté à l'AANN incorporé par une réaction chimique "click". La protéine possède dès lors des propriétés de fluorescence qui permettent de la localiser, d'en étudier les interactions protéine-protéine et d'autres processus. |
|
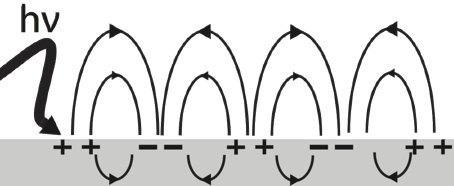
d. Le phénomène de résonance de plasmons de surface (SPR) α. Les plasmons de surface Les plasmons sont des oscillations collectives d'électrons libres, qui existent dans la masse ou à la surface d'un métal ou au voisinage de nanoparticules : les plasmons peuvent ainsi être classés en plasmons de masse, plasmons de surface et plasmons de surface localisée (nanoparticules). La résonance de plasmons de surface ("Surface Plasmon Resonance" - SPR) se produit :
Source : Wikipédia
Exemple de système très utilisée en biologie : le système BIACORE®. |
β. Principe de la SPR appliquée aux molécules biologiques La SPR est donc une méthode optique qui mesure l'indice de réfraction à la surface d'un biocapteur.
Une protéine est immobilisée sur une surface métallique (le biocapteur) et une solution du ligand est injectée sur cette surface.
La SPR nécessite une concentration très faible de matériau (de l'ordre du nM) mais implique l'immobilisation de l'un des partenaires de liaison. [Remarque quant à la terminologie : dans de nombreuses publications et figures, le ligand est dénommé "analyte" et la protéine qui le fixe est dénommée "ligand".] |
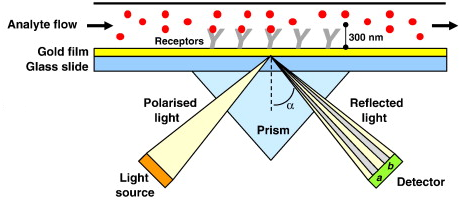
γ. Description du système physique de la SPR La surface est un mince film d'or sur un support en verre qui forme le fond d'une cellule d'analyse de très petit volume (moins de 100 nl) à travers laquelle une solution aqueuse contenant le ligand (le milieu biologique) passe en continu ("flow-cell"). La lumière polarisée monochromatique provenant d'une source laser est dirigée à travers un prisme vers la surface inférieure du film d'or où des plasmons de surface sont générés uniquement sous un angle critique de cette lumière incidente.
Source : Patching S.G. (2014)
Source : Patching S.G. (2014) |
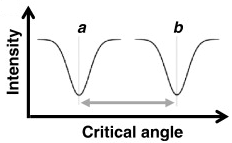
δ. Interprétation d'un sensorgramme SPR
Source : Patching S.G. (2014) Rappel de l'équilibre de fixation d'un ligand L sur une protéine P La vitesse d'association s'écrit : va = ka . [P].[L] - La vitesse de dissociation s'écrit : vd = kd . [PL]
Quand le système est à l'équilibre, les vitesses d'association et de dissociation sont égales :
|
ε. La réponse optimale Rmax (en RU) Elle traduit la capacité de liaison maximale du ligand par la protéine immobilisée : Rmax = (MMligand/MMprotéine) × Rprotéine × nprotéine
SPRD ("Surface Plasmon Resonance Database") : base de données pour optimiser des expériences de SPR. |
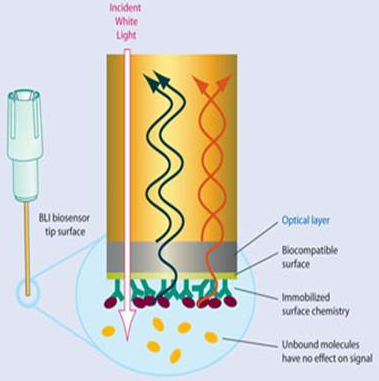
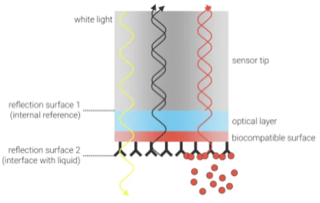
e. L'interférométrie de biocouches α. Principe de la méthode La technique d'interférométrie de biocouches ("Biolayer Interferometry" - BLI) permet de mesurer la cinétique des interactions moléculaires et l'affinité entre biomolécules en analysant le modèle d'interférence de la lumière blanche réfléchie par la pointe d'un biocapteur ("biosensor tip"), fréquemment une fibre optique.
La technique BLI analyse les modèles d'interférence de la lumière réfléchie par 2 couches optiques :
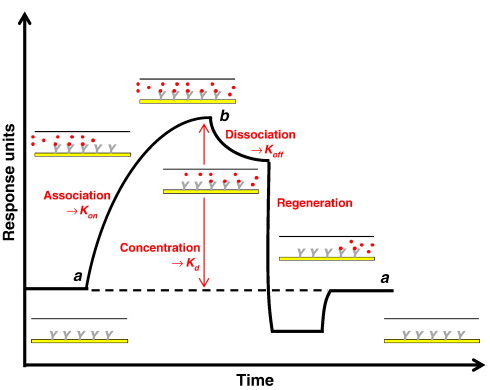
Au fur et à mesure que le ligand se fixe sur la protéine immobilisée, la biocouche est caractérisée par 2 surfaces distinctes : la protéine immobilisée seule et le complexe [protéine immobilisée - ligand].
Source : 2bind Technologies Une lumière blanche ("white light") est projetée sur la pointe du biocapteur et elle est réfléchie par la couche de référence et par la biocouche :
|
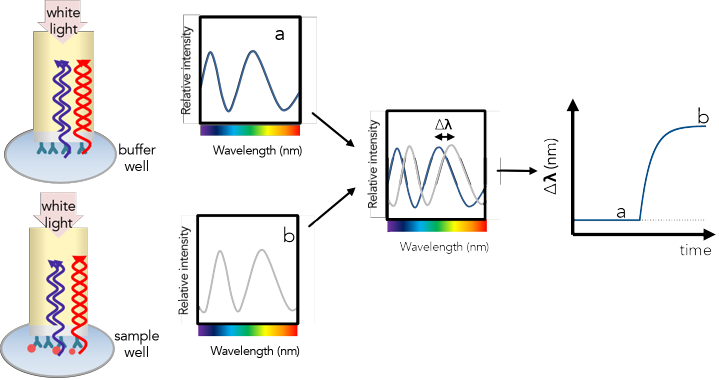
γ. Modèles d'interférence La technique BLI analyse le modèle d'interférence de la lumière blanche lié au changement de la couche externe de la pointe résultant de la fixation du ligand (ou de sa dissociation) : modèle a -> modèle b dans la figure ci-dessous.
Source : CMI - Harvard
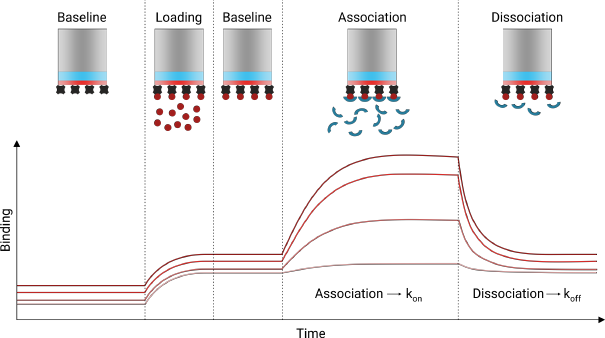
Les mesures de ce changement résolues en temps permettent de déterminer les constantes de vitesse d'association et de dissociation (kon et koff) du ligand avec/de la protéine immobilisée sur la surface de la pointe. β. Caractéristiques importantes de la technique BLI
|
δ. Enregistrement des données d'interférométrie de biocouches La mesure du modèle d'interférence enregistre donc en temps réel (cinétique) les interactions entre molécules. Les capteurs sont déplacés d'un puits d'une microplaque à un autre pour changer le type de solution :
Source : 2bind Molecular interactions
|
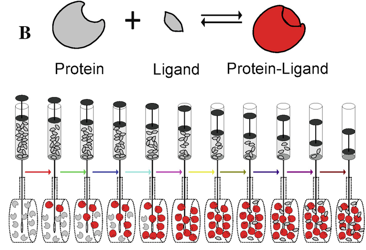
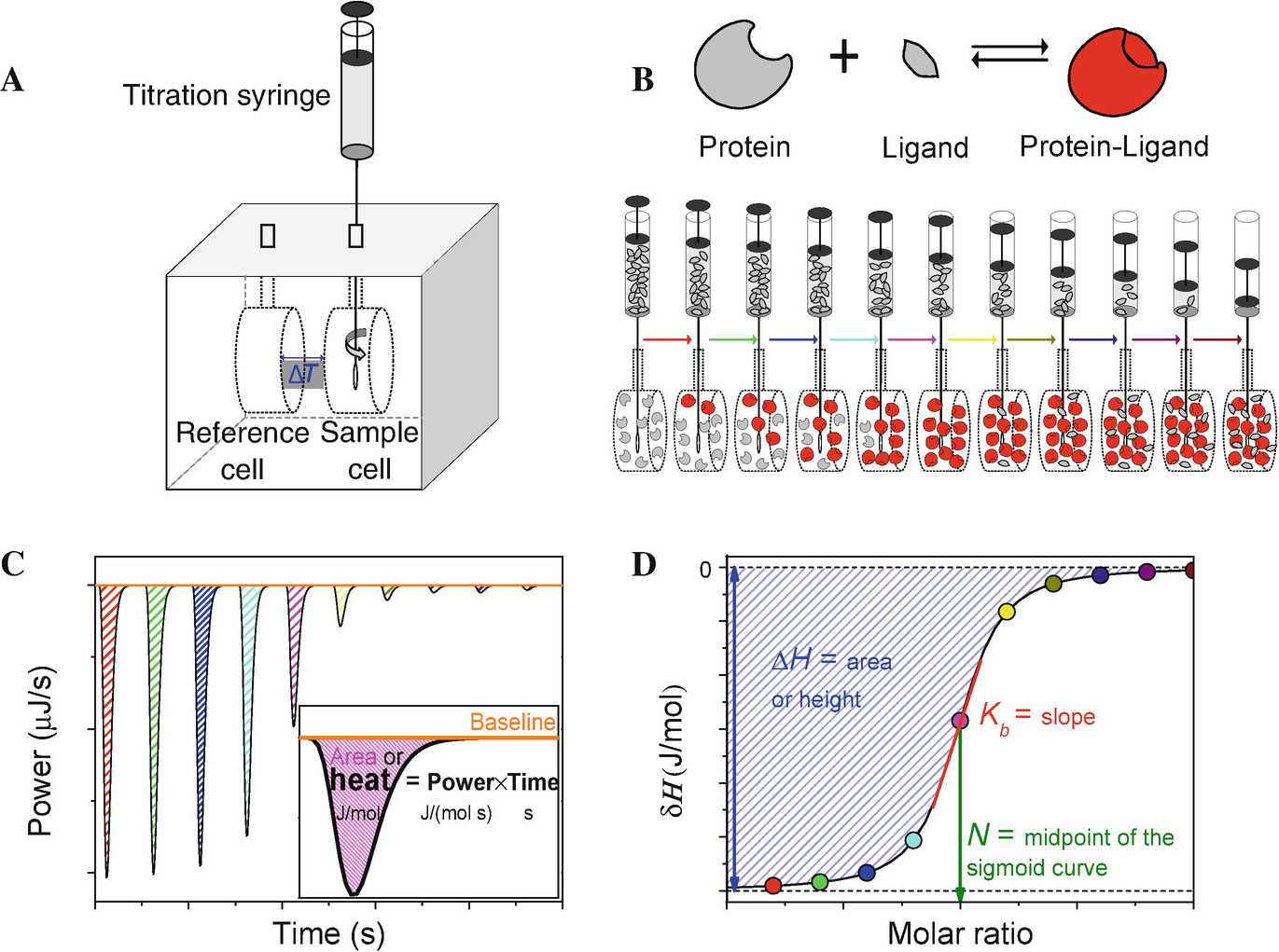
6. Titrage calorimétrique isotherme ("Isothermal Titration Calorimetry" - ITC) L'ITC est une méthode qui permet de mesurer, en théorie, les valeurs quantitatives des grandeurs thermodynamiques liées à l'interaction entre molécules dans leur état natif.
En une seule expérience :
L'ITC est une technique assez complexe qui nécessite une concentration de matériau de l'ordre du μM. |
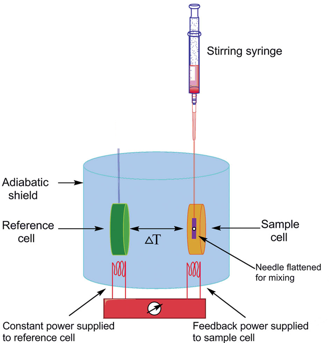
b. Fonctionnement du calorimètre Le calorimètre pour un titrage isotherme est composé de 2 cellules identiques : une cellule de référence ("Reference cell") et une cellule contenant l'échantillon (la protéine par exemple, "Sample cell"). Ces cellules :
Source : Song et al. (2015) Une seringue ("Stirring syringe") est insérée dans la cellule contenant la protéine :
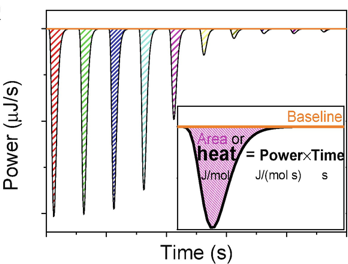
A chaque injection, la réaction protéine + ligand <=> [protéine-ligand] se traduit par une variation d'enthalpie (ΔHréaction) qui s'accompagne d'un dégagement (ou d'une absorption) de chaleur ("heat").
|
|
Au fur et à mesure que les injections du ligand s'accumulent, le rapport molaire [ligand / protéine] augmente et la protéine est progressivement saturée. Bien que minime, puisque les volumes injectés sont très petits, on doit cependant tenir compte de la dilution progressive de la protéine.
Source : Paketuryte et al. (2019) Au fur et à mesure que la protéine est saturée par le ligand, l'amplitude des pics diminue car elle est proportionnelle au nombre de liaisons qui s'établissent.
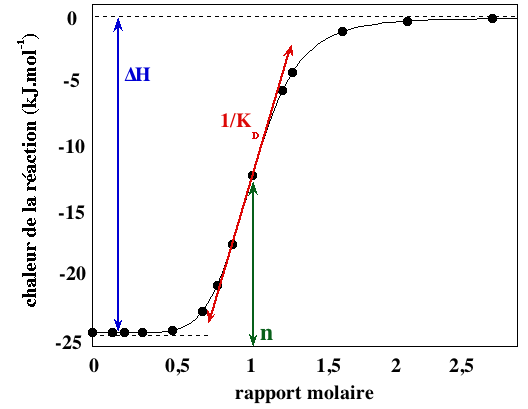
Source : Paketuryte et al. (2019) Quand on a un excès de ligand par rapport à la protéine, la limite inférieure de l'amplitude des pics ne reflète plus que la dilution de la protéine et les effets mécaniques lors d'une nième injection : l'isotherme de fixation est une courbe sigmoïde (voir ci-dessous). Le contenu thermique total dans le volume V0 de la cellule contenant la protéine est : Q = n.[P]0.V0.ΔHréaction.Θ
|
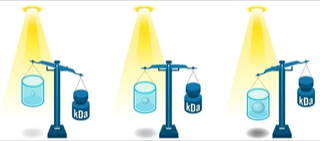
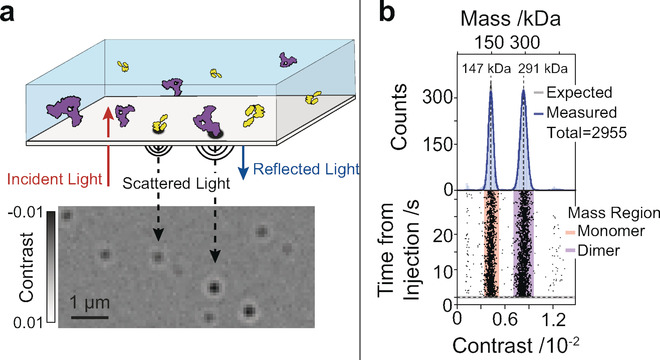
7. La photométrie de masse ("Mass photometry") La photométrie de masse est basée sur les principes de la microscopie à diffusion interférométrique ("interferometric SCATtering microscopy"- iSCAT, Young et al., 2018). C'est est une technique de plus en plus employée pour l'étude des interactions protéine-protéine, les mécanismes d'oligomérisation des protéines et le stabilité des protéines.
Source : REFEYN Lors d'une mesure de photométrie de masse, une protéine en contact avec une surface de mesure est donc éclairée par un faisceau lumineux ("incident light") dans le photomètre de masse (figure ci-dessous).
Source : Soltermann et al. (2020)
L'intensité du signal d'interférence, appelé contraste de photométrie de masse (ou contraste interférométrique), est proportionnelle à la masse molaire des molécules (actuellement jusqu'à 30 KDa, pour une taille inférieure à 100 nm). |
b. Avantages de la photométrie de masse En utilisant un étalon de masse connue et de la même classe de biomolécules que les échantillons analysés, la photométrie de masse permet de déterminer la masse moléculaire de chaque molécule analysée.
Avantages
|
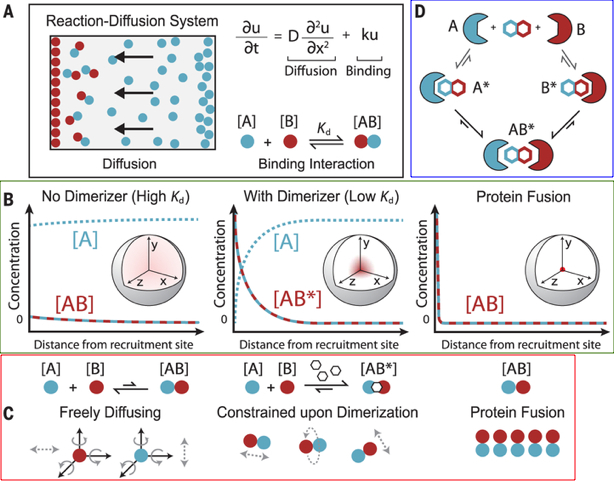
8. Notion de proximité induite chimiquement "Chemically Induced Proximity" a. Modélisation des cinétiques de réactions associées aux mécanismes de proximité induite L'augmentation effective de la concentration en présence d'un inducteur chimique de proximité ("Chemically Induced Proximity" - ICP) sur un site de recrutement peut être décrite en assimilant la dimérisation à une réaction ayant lieu dans un système [réaction-diffusion] (figure A ci-dessous) car les modèles d'équilibre ne peuvent pas décrire des gradients de concentration abrupts.
Source : Stanton et al. (2018) Figures A et B : la variation des concentrations des monomères et des complexes [AB] ou [AB*] dépend du taux de diffusion des molécules et de la vitesse de fixation de la molécule de dimérisation chimique. Ces 2 paramètres dépendent notamment de la distance au site de recrutement (reportée en abscisse sur les graphiques).
Figure C : la minimisation de l'entropie de translation et de rotation sont les principales contributions thermodynamiques à la dimérisation induite chimiquement. |
b. Découverte et développement de petites molécules appelées colles moléculaires Les cibles non "médicamenteuses" ("undruggable targets") désignent des cibles thérapeutiques difficiles à traiter par les approches "traditionnelles". Elles sont caractérisées, par exemple, par l'absence de poches de liaison à des ligands définis, par des modes d'interaction protéine-protéine non catalytiques ou bien encore par des structures 3D peu ou pas résolues. Les travaux pionniers de S. Schreiber et collaborateurs ont permis de développer de nombreuses petites molécules permettant l'interactions entre protéines.
Source : Cully M. (2025) Parmi ces colles moléculaires issues de la recherche, on peut citer les médicaments imides immunomodulateurs ("ImmunoModulatory imide Drugs" - IMiDs) tels que la thalidomide (et ses analogues) qui génèrent des complexes ternaires avec la ligase E3 Cereblon qui entraîne la dégradation spécifique des protéines ciblées par le protéasome. |
La nature suit le concept de colle moléculaire en utilisant des produits naturels qui génèrent tous des complexes ternaires entre protéines cibles fondamentalement différentes. En voici quelques exemples : |
||
| La rapamycine dans le complexe [FKBP12 / rapamycine / FRB] | L'antibiotique macrolide rapamycine forme un complexe avec la protéine cytosolique FKBP12 ("FK506-Binding Protein 12") qui inhibe spécifiquement le complexe TORC1, entraînant un arrêt de la croissance. |
|
FK506 dans le complexe [FKBP12 / FK506 / calcineurine]. |
FK506 est un immunosuppresseur de la cyclosporine A et la calcineurine joue un rôle important dans la virulence fongique et constitue donc une cible antifongique potentielle. | |
| Le produit naturel WDB002 (de la famille FK506 - rapamycine) dans le complexe [FKBP12 / WDB002 / centrosomal protein 250]. | ||
Autres exemples
|
||
c. Exemples de l'implication de la proximité induite chimiquement dans la régulation de processus cellulaires Voir une application in vivo de l'inducteur de proximité chimique mandipropamide ("Mandi").
Autres exemples d'utilisation d'inducteurs de proximité chimique pour l'étude de processus biologiques :
|
9. La prédiction des interactions protéine-protéine par apprentissage profond a. Description des méthodes et des outils informatiques nécessaires Ces méthodes de prédiction sont exclusivement informatiques avec l'utilisation de langages spécifiques du domaine de l'intelligence artificielle. Notamment :
Leurs outils d'apprentissage sont, en particulier, des modèles de langage pré-entraînés pour le traitement du langage naturel ("Natural Language Processing"). Par exemple :
Ces méthodes s'appuient sur des modèles mathématiques sophistiqués. |
Enfin, ces méthodes utilisent des jeux d'entraînement gigantesques (de tailles exponentiellement croissantes) puisés :
Les informations structurales et fonctionnelles des protéines au sein des séquences d'acides aminés sont ainsi apprises automatiquement par des modèles d'apprentissage profond.
Source : Unsal et al. (2022) Ilustration de l'utilisation de grands modèles de langage ("Large Language Models" - LLM) Requêtes (traduites) dans les invites pour GPT-3 (API), GPT-3.5 (ChatGPT) et GPT-4 (ChatGPT) :
Remarque : la dernière phrase de cette requête est étonnante ! |
| Exemples de méthodes basées sur l'analyse des séquences d'acides aminés pour prédire les interactions protéine-protéine | Exemples d'algorithmes & programmes de prédiction des interactions protéine-protéine |
|
|
Voir un cours sur l'apprentissage profond (intelligence artificielle). Voir un cours sur le traitement du langage appliqué aux séquences de protéines. |
|
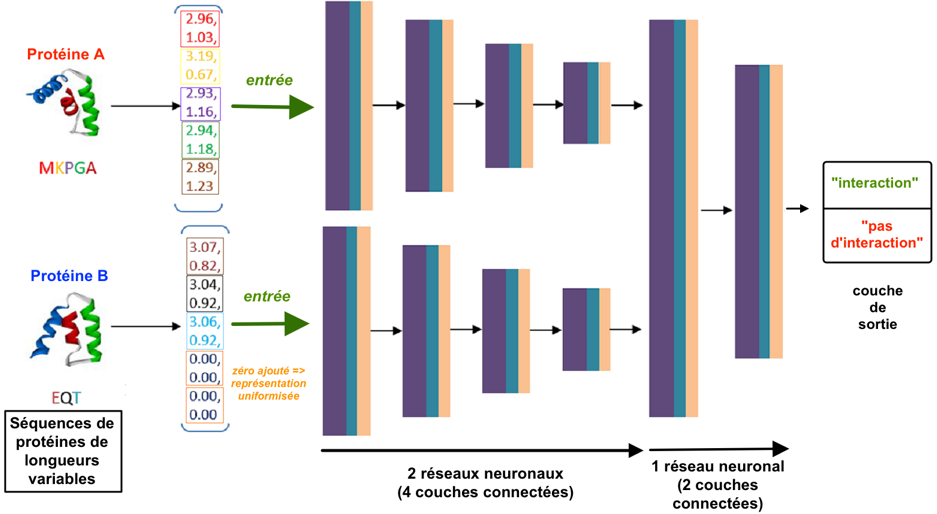
b. Illustration de la méthode "DeepFE-PPI" La longueur de toutes les séquences en acides aminés (composées de D résidus différents) est uniformisée à une valeur fixe m :
Ce modèle est composé de :
Source : Yao et al. (2019)
Les autres caractéristiques de ce modèle d'apprentissage profond sont :
|
Exemples d'autres ressources de prédiction des interactions protéine - protéine DeepPPAPred : outil de prédiction d'affinité de liaison pour les protéines basé sur l'apprentissage profond. DDMut-PPI : serveur Web qui prédit par une méthode d'apprentissage profond les effets de mutations ponctuelles sur les interactions protéine-protéine. |
| 10. Liens Internet et références bibliographiques | |
Cours en ligne "Protein-protein interactions" Pathway Figure OCR : extraction d'informations publiées dans la littérature. Pathway Commons Ensemble d'articles scientifiques récents utilisant la méthode SPR pour diverses protéines |
|
PPI-MASS: An Interactive Web Server to Identify Protein-Protein Interactions From Mass Spectrometry-Based Proteomics Data The working principle of isothermal titration calorimetry SPRD : base de données pour optimiser des expériences de SPR. Photométrie de masse Site "Molecular glues tackle undruggable targets" |
|
Smith & Johnson (1988) "Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase" Gene 67, 31 - 40 Fields & Song (1989) "A novel genetic system to detect protein-protein interactions" Nature 340, 245 - 246 Guan & Dixon (1991) "Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase" Anal. Biochem. 192, 262 - 267 Spencer et al. (1993) "Controlling signal transduction with synthetic ligands" Science 262, 1019 - 1024 Pierce et al. (1999) "Isothermal titration calorimetry of protein-protein interactions" Methods 19, 213 - 221 |
|
Zhu et al. (2001) "Global analysis of protein activities using proteome chips" Science 293, 2101 – 2105 Turnbull et al. (2003) "On the value of c: can low affinity systems be studied by isothermal titration calorimetry ?" J. Am. Chem. Soc. 125, 14859 - 14866 Chatr-aryamontri et al. (2007) "MINT: the Molecular INTeraction database" Nucleic Acids Res. 35, D572 - D574 |
|
Reynolds et al. (2012) "Multivalent gold glycoclusters: high affinity molecular recognition by bacterial lectin PA-IL" Chemistry 18, 4264 - 4273 Kodama & Hu (2012) "Bimolecular fluorescence complementation (BiFC): a 5-year update and future perspectives" Biotechniques 53, 285 - 298 Rhee et al. (2013) "Proteomic Mapping of Mitochondria in Living Cells via Spatially-Restricted Enzymatic Tagging" Science 339, 1328 - 1331 Patching S.G. (2014) "Surface plasmon resonance spectroscopy for characterisation of membrane protein-ligand interactions and its potential for drug discovery" Biochim. Biophys. Acta. 1838, 43 - 55 Kohl et al. (2014) "Ultrafast tissue staining with chemical tags" Proc. Natl. Acad. Sci. USA 111, E3805 - E3814 Song et al. (2015) "Choosing a suitable method for the identification of replication origins in microbial genomes" Front. Microbiol. 6, 1049 Mehla et al. (2015) "The yeast two-hybrid system: a tool for mapping protein-protein interactions" Cold Spring Harb. Protoc. (5), 425 - 430 |
|
Xing et al. (2016) "Techniques for the analysis of protein-protein interactions in vivo" Plant Physiol. 171, 727 - 758 Fernandes et al. (2016) "Systematic analysis of the gerontome reveals links between aging and age-related diseases" Hum. Mol. Genet. 25, 4804 - 4818 Hu et al. (2017) "Functional Analysis of Human Hub Proteins and Their Interactors Involved in the Intrinsic Disorder-Enriched Interactions" Int. J. Mol. Sci. 18, 2761 Di Silvestre et al. (2018) "Large Scale Proteomic Data and Network-Based Systems Biology Approaches to Explore the Plant World" Proteomes 6, 27 Stanton et al. (2018) "Chemically induced proximity in biology and medicine" Science 359, eaao5902 |
|
|
Ivarsson & Jemth (2019) "Affinity and specificity of motif-based protein-protein interactions" Curr. Opin. Struct. Biol. 54, 26 - 33 Paketuryte et al. (2019) "Inhibitor Binding to Carbonic Anhydrases by Isothermal Titration Calorimetry" dans "Carbonic Anhydrase as Drug Target" pages 79 - 95, Springer Yao et al. (2019) "An integration of deep learning with feature embedding for protein–protein interaction prediction" PeerJ. 7, e7126 Lee et al. (2019) "Site-specific labeling of proteins using unnatural amino acids" Mol. Cells 42, 386 - 396 Soltermann et al. (2020) "Quantifying Protein–Protein Interactions by Molecular Counting with Mass Photometry" Angew. Chem. Int. Ed. Engl. 59, 10774 - 10779 |
|
Soltermann et al. (2021) "Label-free methods for optical in vitro characterization of protein–protein interactions" Phys. Chem. Chem. Phys. 23, 16488 - 16500 Olerinyova et al. (2021) "Mass Photometry of Membrane Proteins" Chem. 7, 224 - 236 Shandell et al. (2021) "Genetic code expansion: a brief history and perspective" Biochemistry 60, 3455 - 3469 Laxman et al. (2021) "The benefits of unnatural amino acid incorporation as protein labels for single molecule localization microscopy" Front. Chem. 9, 641355 |
|
Unsal et al. (2022) "Learning functional properties of proteins with language models" Nat. Mach. Intell. 4, 227 - 245 Lee M. (2023) "Recent Advances in Deep Learning for Protein-Protein Interaction Analysis: A Comprehensive Review" Molecules 28, 5169 Rehana et al. (2023) "Evaluation of GPT and BERT-based models on identifying protein-protein interactions in biomedical text" arXiv:2303.17728 Rui et al. (2023) "Protein–protein interfaces in molecular glue-induced ternary complexes : classification, characterization, and prediction" RSC Chem. Biol. 4, 192 - 215 |
|
Newcombe et al. (2024) "Stereochemistry in the disorder–order continuum of protein interactions" Nature 636, 762 - 768 Ren et al. (2024) "Comprehensive Review on Bimolecular Fluorescence Complementation and Its Application in Deciphering Protein–Protein Interactions in Cell Signaling Pathways" Biomolecules 14, 859 Zhou et al. (2024) "DDMut-PPI: predicting effects of mutations on protein-protein interactions using graph-based deep learning" Nucleic Acids Res. 52(W1), W207 - W214 Nikam et al. (2024) "DeepPPAPredMut: deep ensemble method for predicting the binding affinity change in protein–protein complexes upon mutation" Bioinformatics 40, btae309 Budiarta et al. (2024) "Site-specific protein labeling strategies for super-resolution microscopy" Curr. Opin. Chem. Biol. 80, 102445 |
|
Cully M. (2025) "Natural molecular glues spur next-gen small molecules for 'undruggable' targets" Nat. Biotechnol. 43, 1212 - 1214 Repity et al. (2025) "Nondegradative Synthetic Molecular Glues Enter the Clinic" ChemMedChem 20, e202500048 Chen et al. (2025) "Target sequence-conditioned design of peptide binders using masked language modeling" Nat. Biotechnol. doi: 10.1038/s41587-025-02761-2 de Luis et al. (2025) "Fluorescent labeling of proteins in vitro and in vivo using encoded peptide tags" J. Biol. Chem. 301, 110229 Dakhnevich et al. (2025) "Pyrrolysine aminoacyl-tRNA synthetase as a tool for expanding the genetic code" Int. J. Mol. Sci. 26, 539 |
|
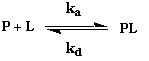
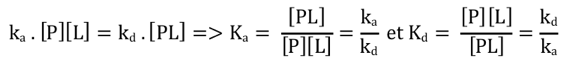

{kind=link}
{kind=link}