| Quelques méthodes d'étude de la cellule et des macromolécules biologiques |
| Tweet |
|
|
1. Synopsis général des techniques 2. L'adhésion entre cellules (desmosome, cadhérines) 3. La technique de cryofracture 4. Les techniques de centrifugation 5. Les techniques immunochimiques 6. Les techniques de fluorescence pour l'hybridation in situ 7. Les structures des macromolécules biologiques et certaines techniques pour les étudier
|
8. Synthèse des protéines 9. Dégradation des protéines - ubiquitine et protéasomes 10. Liens Internet et références bibliographiques |
|
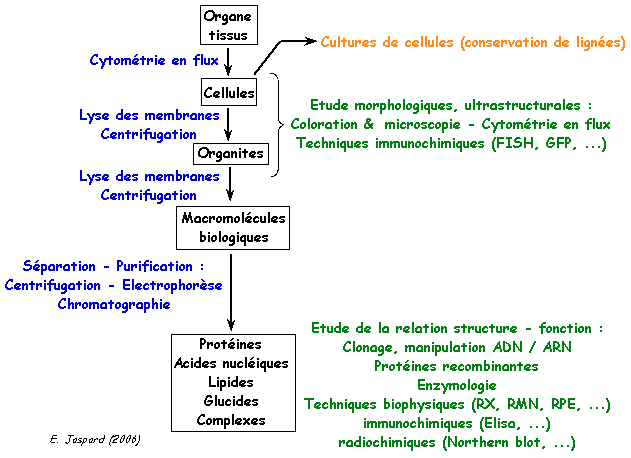
1. Synopsis général des techniques pour aller de l'observation des tissus à la purification et l'analyse des macromolécules biologiques
|
| Techniques (liste non exhaustive) | But |
| coupes de tissus (microtome) - isolement de cellules | prélèvement |
| coloration - histologie - immunohistochimie | observation microscopique - cytologie |
| cytométrie en flux | tri des cellules - observation de différents marqueurs phénotypiques - apoptose |
| culture cellulaire | multiplication des cellules - sauvegarde des cellules |
| immunohistochimie | localisation intracellulaire - mise en évidence d'organites, de macromolécules typiques |
| lyse des cellules (détergents, chlorure guanidinium, lysozyme, ...) | rupture de la membrane plasmique - extraction de macromolécules ou d'organites |
| ultracentrifugation en gradient de densité (saccharose) | séparation des cellules en fonction de la constante de sédimentation (Svedberg) |
| ultracentrifugation différentielle | séparation et purification des organites |
| techniques de chromatographie (échange d'ions, gel de filtration, affinité, ...) | séparation et purification des macromolécules biologiques (surtout protéines) |
| immunocytochimie (ELISA, EIA, "radioimmunoassay", ...) | localisation cellulaire des macromolécules biologiques |
| electrophorèse sur gel d'agarose | analyse de l'ADN (tailles et formes des fragments) |
| electrophorèse sur gel de polyacrylamide en conditions natives ou dénaturantes (SDS) | analyse des protéines (masse molaire / nombre de sous-unité / structure quaternaire) |
| méthodes de dosage spectrophotométrique (absorption à 280 nm, méthode de Bradford, méthode de Lowry ...) | détermination de la concentration des protéines |
| dosage enzymatique | mesure de l'activité catalytique des enzymes |
| construction de banques et clonage d'ADNc | surexpression de protéines recombinantes |
cristalographie - diffraction des rayons X et résonance magnétique nucléaire |
détermination de la structure tridimensionnelle des macromolécules |
| cryo-microscopie électronique | détermination de la structure tridimensionnelle des complexes supra-moléculaires (exemples : ribosome, protéasome), d'organites, de virus |
| Bioinformatique | prédiction de structures (primaire, secondaire, tertiaire), de motifs impliqués dans la fonction des macromolécules ou leur interaction avec d'autres entités biologiques, ... |
| Visualisation du réticulum endoplasmique en 3D. | |
|
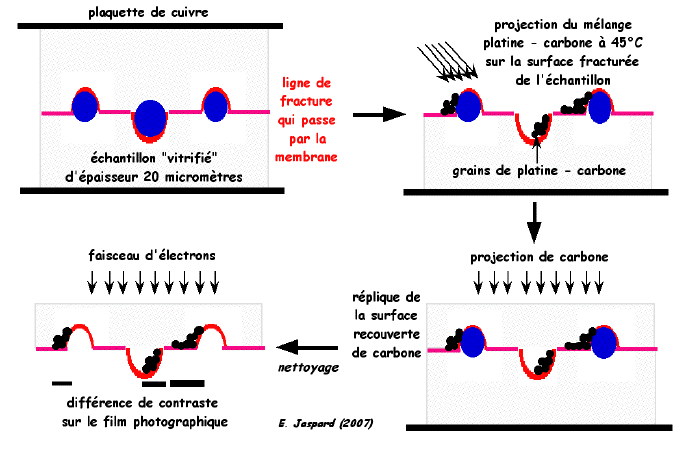
3. La technique de cryofracture Principe La cryofracture révèle la localisation des protéines membranaires. En effet, selon qu'elles sont intrinsèques ou extrinsèques, volumineuses ou pas, les protéines restent attachées à une face de la membrane ou à une autre. La fracture suit le milieu de la bicouche lipidique qui est la région de la membrane offrant le moins de résistance. Technique
Le carbone est traversé par les électrons mais le platine les arrête plus ou moins totalement selon l'épaisseur rencontrée par le faisceau d'électrons. Un film photographique traduit les informations sur le relief en contraste de gris :
Source : "Cryofracture" (CNRS) |
|
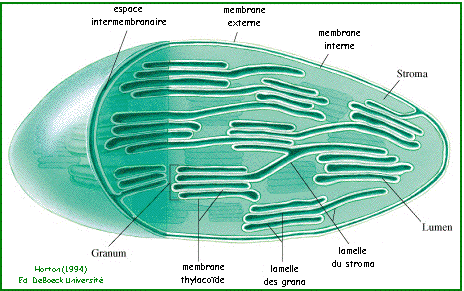
Application à l'étude du chloroplaste Les chloroplastes ont deux membranes (interne et externe) bordant une zone aqueuse appelée stroma (siège de la phase sombre de la photosynthèse). Le stroma contient la membrane thylacoïde (siège de la phase diurne). La membrane thylacoïde est plissée en un réseau de nombresuses vésicules aplaties qui prend la forme :
Les parties de membrane thylacoïde :
L'espace interne enclos par la membrane thylacoïde est le lumen.
Source : "Principes de Biochimie" Horton et al. (1994) Comparer cette figure "théorique" aux clichés obtenus par cryofracture du site : "La structure du chloroplaste" - Equipe multimédia - Université Jussieu |
| produit | densités | avantage | inconvénient |
| saccharose | 1,3 g/mL (2,5 M) | peu coûteux / électriquement neutre | problème de pression osmotique : à éviter pour l'isolement de cellules entières |
| chlorure de césium (CsCl) | 1,9 g/mL (7,5 M) | densité très élevé trés adapté pour la séparation des acides nucléiques | coût / sel |
| Metrizamide (dérivé iodobenzamido du glucose) | 1,5 g/mL | très soluble et non chargé | coût / forte absorption dans l'UV / instabilité |
| Ficoll (polymère de glucose) | --- | densités élevées sans générer de fortes pressions osmotiques adapté à la séparation de cellules entières | coût / forte viscosité |
| Percoll (silice recouverte de polyvinylpyrolidone) | 1,3 g/mL | faibles pressions osmotiques et faible viscosité adapté à la séparation de cellules entières | coût / faibles densités atteintes |
|
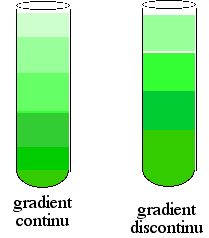
La vitesse de sédimentation peut donc être modifiée en faisant varier de façon continue ou discontinue (par paliers) cette différence en créant un gradient de concentration du milieu ambiant.
Source : "Centrifugation en gradient de densité" |
|
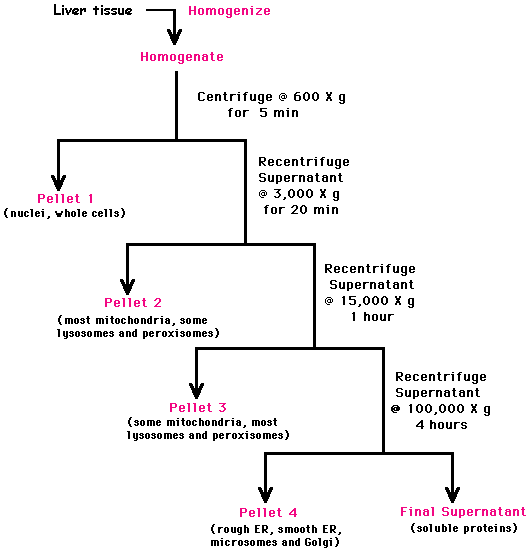
b. Centrifugation différentielle Elle permet de séparer les particules (organites, macromolécules, ...) en fonction de leur taille par une succession de centrifugations à des temps et des accélérations croissants. Figure ci-dessous : protocole de purification de différents organites de la cellule.
Source : "Purification by Differential Centrifugation" |
|
5. Les techniques immunochimiques Le but de l'immunochimie est de révéler une molécule biologique présente sur une cellule ou un tissu avec des anticorps spécifiques.
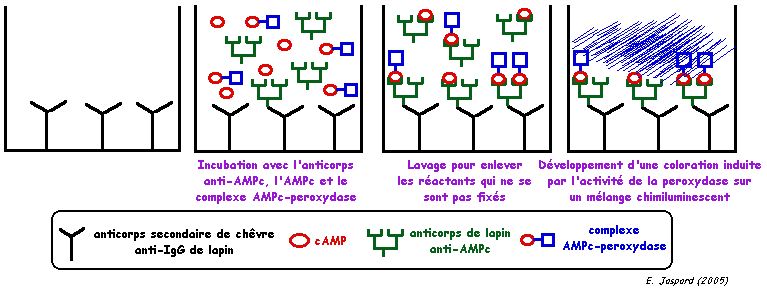
Principe : un anticorps primaire est fixé sur un antigène. On révèle cet anticorps avec un second anticorps dirigé contre la classe d'immunoglobuline à laquelle appartient l'anticorps primaire. Des particules d'or ("immunogold") ou une enzyme qui catalyse une réaction colorimétrique (exemple : la peroxydase) sont fixées sur l'anticorps secondaire. Ci-dessous, schéma d'un dosage immuno-enzymatique ("enzyme-linked immunoassay" ou EIA).
Le milieu de développement de la chimiluminescence contient du luminol (3-aminophthalhydrazide), du 4-iodophenol et de l'eau oxygénée. Des radicaux libres sont formés par l'action de la peroxydase sur l'eau oxygénée. Ces radicaux libres réagissent avec le luminol qui émet alors des photons de fluorescence de longueur d'onde 425 - 445 nm. Des techniques de couplage à l'enzyme de révélation permettent d'augmenter la sensibilité du dosage en diminuant la limite de détection. L'une d'entre elles consiste à fixer plusieurs molécules de biotine sur l'anticorps. La biotine est ensuite reconnue par la streptavidine (sur laquelle est fixée la peroxydase) avec une trés haute affinité. |
|
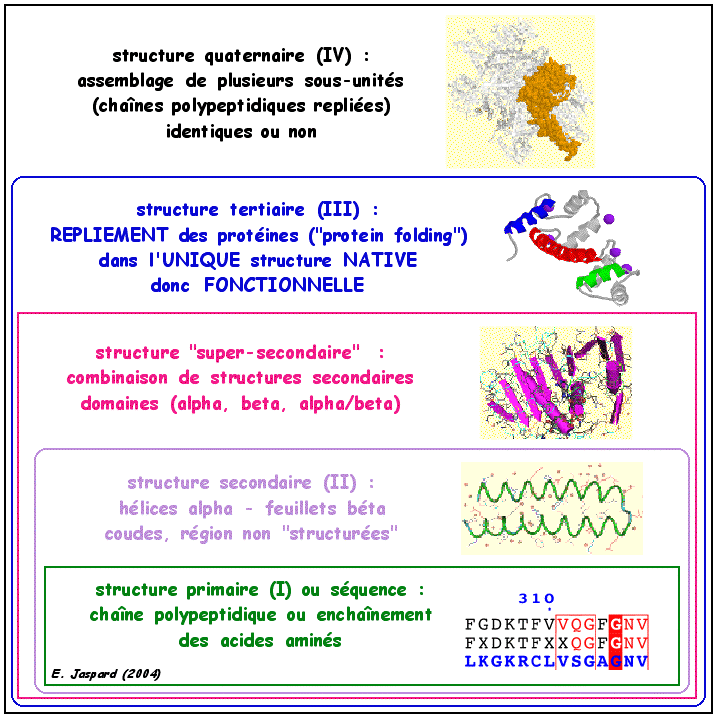
7. Les structures des macromolécules biologiques et les techniques pour les étudier Il existe quatre niveaux de structure des protéines :
Toutes les protéines ne sont pas structurées dans leur conformation native. Un très grand nombre sontdites intrinsèquement non structurées. |
|
a. Conditions de lyse de bactéries pour la purification d'une protéine recombinante (DEA : B. Saleh - 2000) Les souches E. coli B834(DE3)pLysS sont cultivées à 37°C sur milieu Luria Bertani (LB) en présence d'antibiotiques : carbénicilline plus chloramphénicol (la résistance au chloramphénicol étant conférée par le plasmide pLysS). L'expression de la protéine recombinante est induite par l'addition d'isopropylthiogalactoside (IPTG) quand la culture des bactéries atteint une densité optique A600 = 0,41. Les bactéries sont centrifugées 15 min à 6000g et le culot bactérien est resuspendu dans un tampon de lyse constitué de : Tris-HCl 50 mM, pH 7,5 ; EDTA 1 mM ; PMSF 1 mM ; NaCl 100 mM ; sarcosine 0,2% ; lysozyme 0,25 mg/mL ; DNase I 60 unités. Ce mélange est incubé 2 heures à 37°C puis le lysat est centrifugé 40 min à 15000 g. Rôle des composants du milieu de lyse Le but est de déstructurer la membrane bactérienne.
Source : "Principes de Biochimie" Horton et al. (1994)
Voir "Solubilization of integral membrane proteins by detergents" (Figure 12.4) au NCBI. Une conséquence de la transformation de bactéries par un vecteur d'expression qui possède un promoteur fort, mais également du caractère réducteur du cytoplasme (le rapport glutathion forme réduite / forme oxydée est estimé entre 30:1 chez les procaryotes et 100:1 chez les eucaryotes), est l'accumulation de certaines protéines recombinantes dans des corps d'inclusion. Figure ci-dessous : cellule d'E. coli 6 heures après l'induction de l'expression de la chaîne lourde de l'anticorps MAK33. Le corps d'inclusion est la structure amorphe en gris clair, à droite.
Source : Lilie et al. (1998) Curr. Opin. Biotech. 9, 497 - 501 |
|
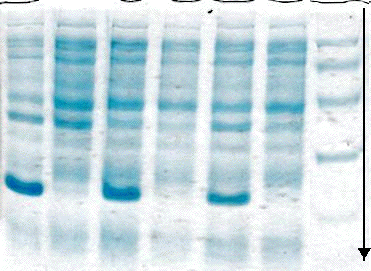
b. Gel d'électrophorèse en conditions dénaturantes Le principe du gel d'électrophorèse en conditions dénaturantes ("sodium dodecyl sulfate polyacrylamide gel electrophoresis", SDS-PAGE) est de traiter un mélange de protéines avec :
Les protéines sont donc sous une forme monomérique. Les protéines de l’échantillon sont ensuite séparées par une électrophorèse sur gel de polyacrylamide à un certain pourcentage. La migration s'effectue sous l'action d'un champ électrique (la flèche de la figure ci-dessous indique le sens de migration).
Une protéine migre d'autant moins que sa masse molaire est élevée. Les protéines sont fixées dans le gel est révélées par une coloration (exemple : le bleu de Coomassie ou le nitrate d'argent). On obtient les différentes bandes de chaque piste de la figure ci-contre. La masse molaire des protéines est déterminée à l'aide de marqueurs qui sont des protéines standards de masses molaires connues (piste de droite) : Exemple : phosphorylase β (97 kDa), albumine (66 kDa), ovalbumine (45 kDa), anhydrase carbonique (30 kDa), inhibiteur de trypsine (20,1 kDa), α lactalbumine (14,4 kDa). Cette technique permet de :
|
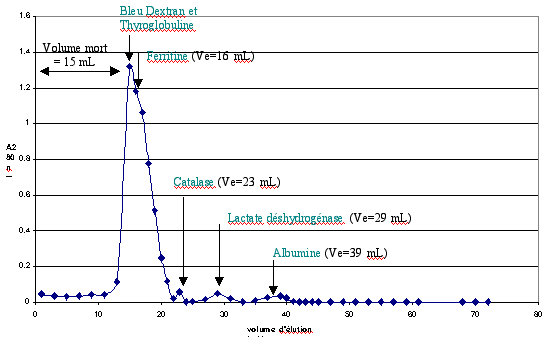
c. Chromatographie de gel de filtration ou tamis moléculaire ou d'exclusion stérique Ce type de chromatographie permet de séparer les protéines et de déterminer leurs masses molaires. Pour celà, il faut calibrer le gel et obtenir une droite de calibration. Un mélange de protéines standards de masses molaires connues sont séparées (figure ci-dessous) dans les mêmes conditions que les protéines de l’échantillon. Exemple : thyroglobuline (669 kDa), ferritine (440 kDa), catalase (232 kDa), lactate déshydrogénase (140 kDa) et albumine (66 kDa).
Le profil d’élution est tracé après mesure d’absorbance à 280 nm des fractions éluées.
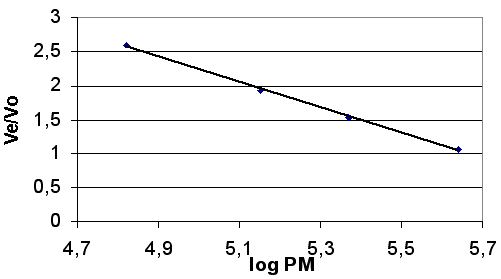
La droite de calibration est : Ve/Vo = f (log PM) (figure ci-dessous).
Les protéines de l’échantillon sont ensuite séparées dans les mêmes conditions et l’absorbance à 280 nm des fractions éluées est mesurée. Par comparaison avec la droite étalon, on peut ainsi déterminer la masse molaire des protéines. |
|
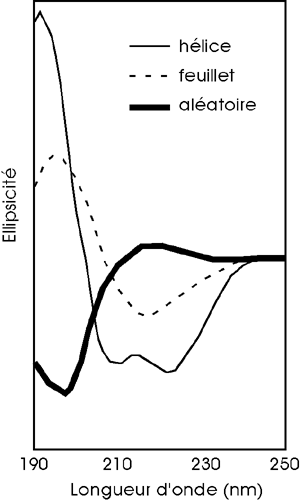
Le dichroïsme circulaire s'appuie sur la capacité des molécules qui ont une activité optique : propriété d'absorber différemment la lumière polarisée (circulairement à droite et circulairement à gauche). Le dichroïsme circulaire est une technique qui permet d'analyser le contenu en structures secondaires des protéines ou des acides nucléiques. Cette une technique non destructive qui permet d'étudier les changements de conformation des protéines dans différents environnements (pH, agents dénaturants, température). Pour déterminer la proportion de chaque type de structure secondaire, il faut effectuer une déconvolution en composantes élémentaires du spectre de dichroïsme circulaire avec des logiciels appropriés.
La forme du spectre de dichroïsme circulaire obtenu pour une protéine peut-être le suivant :
Chaque composante donne une courbe spectrale caractéristique. |
|
e. Détermination de la structure tridimensionnelle par cristallographie et diffraction des rayons X La cristallographie est la science qui se consacre à l'étude des substances cristallines à l'échelle atomique. L'état cristallin est défini par un caractère périodique et ordonné à l'échelle atomique ou moléculaire (maille élémentaire). La cristallogénèse est la formation d'un cristal, soit en milieu naturel, soit de façon expérimentale. C'est le passage d'un état désordonné liquide à un état ordonné solide, contrôlé par la température, la pression, le temps d'évaporation et des lois cinétiques complexes. Les macromolécules biologiques sont souvent très difficiles à cristalliser. C'est par cristallographie que J. Watson, F. Crick, M. Wilkins et R. Franklin ont pu déterminer la structure en double hélice de l'ADN en 1953 (Prix Nobel en 1962).
|
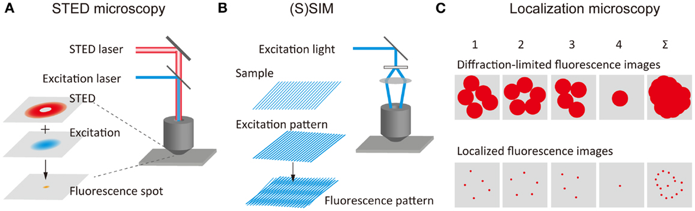
f. Nano-microscopie à fluorescence Les lauréats du prix Nobel de chimie 2014 (Eric Betzig, Stefan Hell et William Moerner) ont développé deux méthodes qui, combinées, permettent de contourner les limites physiques de la microscopie optique (estimée jusqu'à lors à environ 0,2 microns).
Betzig, Hell et Moerner ont développé la microscopie à fluorescence à très haute résolution ("Super-resolved fluorescence microscopy") en utilisant des molécules fluorescentes.
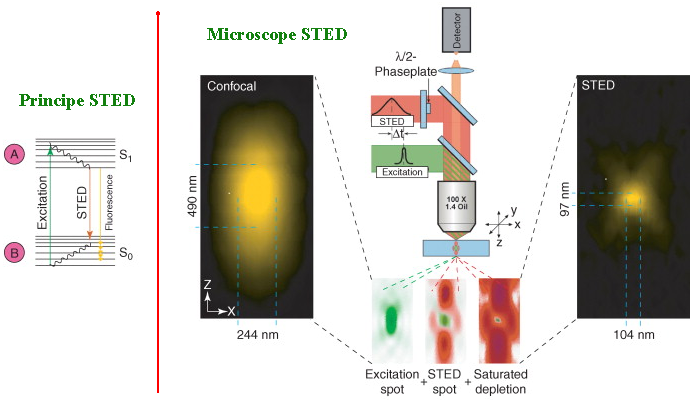
Source : S. Habuchi (2014) Stefan Hell : "Stimulated Emission Depletion" Deux impulsions de rayons laser sont utilisés dans un intervalle de temps très court :
La différence entre les deux images obtenues élimine le bruit de fond lumineux et permet d'obtenir l'image à l'échelle du nanomètre.
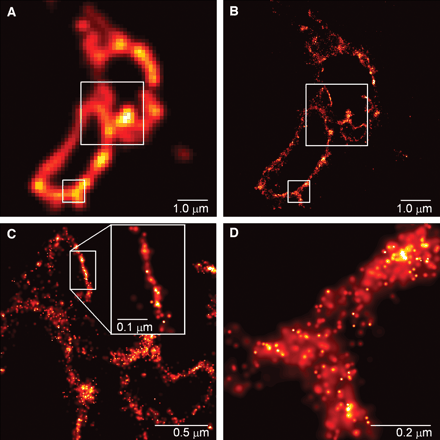
Source : S. Hell et al. (2004) Eric Betzig et William Moerner : "Single-Molecule Microscopy" Technique qui mesure l'absorption lumineuse d'une seule molécule. Une molécule située au voisinage de la GFP ("Green Fluorescent Protein") illuminée à 405 nanomètres est "allumée" et "éteinte" à volonté.
Source : Betzig et al. (2006) |
| 10. Liens Internet et références bibliographiques | |
|
"Biologie cellulaire : exercices et méthodes" Thiry et al. (2014) - DUNOD - ISBN : 978-2-10-070711-9 "Principes de Biochimie" Horton et al. (1994) - De Boeck Université - ISBN : 2-8041-1578-X J.-C. CALLEN - "Biologie Cellulaire" (1999), Ed. DUNOD - ISBN : 2 10 003197 X |
|
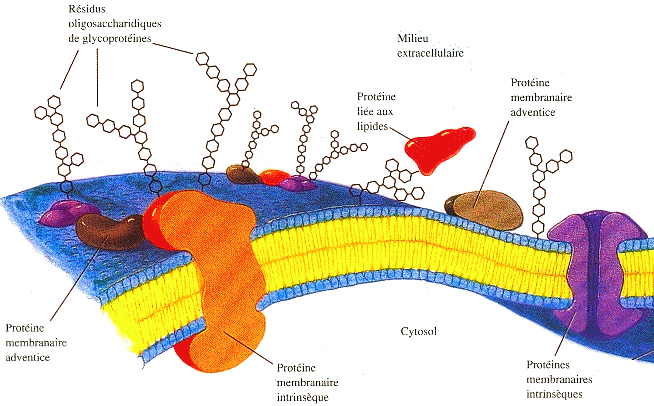
The Cell Image Library Cours très complet au NCBI : "Structure of the Plasma Membrane" |
|
|
Lilie et al. (1998) "Advances in refolding of proteins produced in E. coli" Curr. Opin. Biotech. 9, 497 - 501 Dickson et al. (1997) "On/off blinking and switching behaviour of single molecules of green fluorescent protein" Nature 388, 355-358 S. Hell et al. (2004) "Concepts for nanoscale resolution in fluorescence microscopy" Curr. Opin. Neurobiol. 14, 599–609 Betzig et al. (2006) "Imaging Intracellular Fluorescent Proteins at Nanometer Resolution" Science 313, 1642-1645 S. Habuchi (2014) "Super-resolution molecular and functional imaging of nanoscale architectures in life and materials science" Front. Bioeng. Biotechnol. |
|
![]()