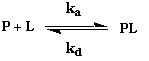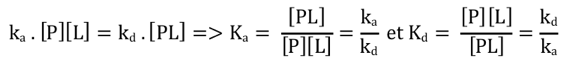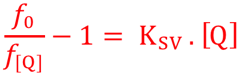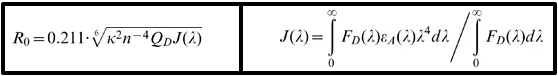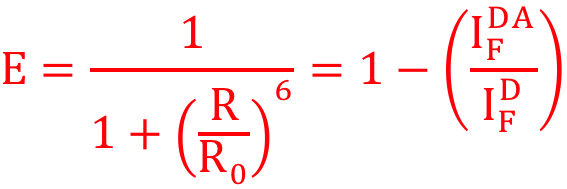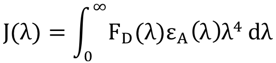
| Caractéristiques et moyens d'étude des interactions entre macromolécules biologiques |
Préambule 1. La relation structure - fonction des macromolécules biologiques 2. Identification et quantification des protéines a. Les protéomes et la protéomique 3. Notions d'interactomes 4. Quantification de l'interaction entre macromolécules biologiques a. Constantes d'équilibre d'association KA et de dissociation KD 5. Les protéines ou régions intrinsèquement désordonnées (IDP/IDR) a. Introduction et définitions 6. Quelques méthodes pour prouver les interactions protéine-protéine a. La technique de complémentation double-hybride |
7. La purification par chromatographie d'affinité couplée à la spectromètrie de masse ("AP-MS") 8. Application de la fluorescence à l'analyse des macromolécules biologiques
9. La technique du FRET appliquée aux molécules biologiques
10. Le FRET appliquée à l'étude de molécule unique ("single-molecule FRET") a. Introduction 11. La technique FISH 12. Apprentissage profond et prédiction des interactions protéine-protéine 13. Liens Internet et références bibliographiques |
Préambule Les interactions entre molécules au sein de la cellule et à la surface de la cellule sont l'élément clé de tous les processus cellulaires, donc de la vie.
En conséquence, il n'est pas un évènement qui se déroule dans une cellule ou à sa surface (reconnaissance et communication, réactions biochimiques, voie de signalisation, transports, processus plus globaux, ...) qui ne mette en jeu la rencontre (collision) suivie d'une éventuelle interaction entre molécules. Cette rencontre est contrôlée notamment par de nombreux paramètres et/ou "acteurs". |
a. Les molécules d'eau dans les cellules Rappel : l'eau "libre" a une concentration de 55,5 M (masse d'1 L H20 = 1000 g / masse molaire (H20) = 18 g.mol-1). Rappel du Nombre d'Avogadro ≈ 6,022 × 1023 mol-1 L'eau représente 60 à 75 % du poids du corps humain. Elle est présente à l'intérieur (environ 2/3 des molécules) et à l'extérieur (environ 1/3 des molécules) des cellules d'un mammifère. La distance moyenne entre les macromolécules dans le cytoplasme est d'environ 1 nm, soit 3 à 4 couches de molécules d'eau. L'eau liquide forme un réseau dynamique de liaisons hydrogène, chaque liaison ayant une durée de vie moyenne d'environ 1 ps (10-12 s). Les molécules d'eau sont donc en mouvement permanent (liaison H établie / rompue) et contribuent à l'hydratation et la diffusion des molécules biologiques. Il est capital de mentionner que le cytoplasme est dans un état physique qui s'apparente davantage à du "verre liquide" aux propriétés hydrodynamiques très différentes de l'eau liquide. |
b. La concentration très élevée des molécules biologiques Gamme de concentrations : 100 à 450 mg.mL-1 (5 - 40 % du volume cytoplasmique) / 300 à 400 mg.mL-1 chez E. Coli. Or la concentration régit la vitesse des molécules donc la fréquence de leurs collisions efficaces (celles qui débouchent sur l'action attendue de cette rencontre).
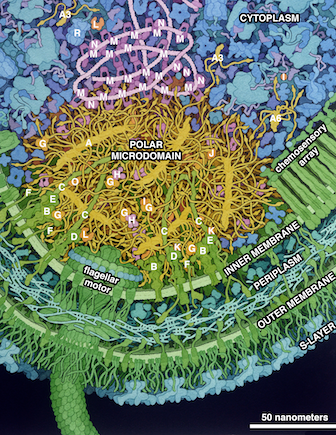
Figure ci-dessus : modèle d'un cytoplasme bactérien composé de protéines, d'ARN, de métabolites, d'ions et d'eau. |
| Quelques chiffres clés de la cellule | ||
| Type de molécule | Quantité dans 1 cellule | Type de cellule |
| Une concentration de 1 nM correspond à | ≈ 1 molécule | Escherichia coli |
| ≈ 1000 molécules | Hela | |
| Une concentration de 100 mg/ml correspond à | ≈ 106 - 107 molécules | Escherichia coli |
| Nombre de protéines Nombre de métabolites Nombre de lipides |
3 106 - 4 106 3 108 2 107 |
Escherichia coli |
| Nombre d'ARNm | 15 103 - 40 103 | Saccharomyces cerevisiae |
| Nombre de protéines "TATA Binding Protein" Nombre de protéines LipL30 Nombre de facteurs de transcription AHR |
20 103 40 103 12 103 |
Saccharomyces cerevisiae Leptospira interrogans lignée cellulaire de fibroblastes murins NIH 3T3 |
| Source : B10NUMB3R5 ("Bionumbers", Harvard) | ||
c. Les entités subcellulaires (membranes, cytosquelette, ...) qui délimitent et confèrent leur structure à certaines cellules. d. La compartimentation de certaines cellules en organites :
|
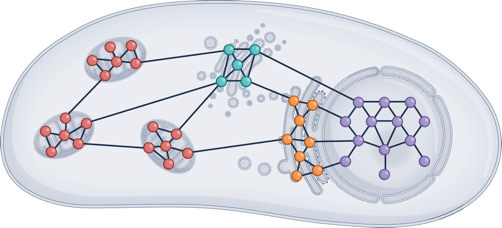
e. Les condensats biomoléculaires ("biomolecular condensates") au sein de quasiment tous les types de cellules (procaryotes et eucaryotes) :
Source : Goodsell & Lasker (2023)
|
1. La relation structure - fonction des macromolécules biologiques a. Les différents types de ligands La molécule qui se fixe sur une autre macromolécule biologique est appelée ligand de manière générique. Un ligand peut être n'importe quel type de molécule biologique :
|
b. Propriétés physico-chimiques des macromolécules biologiques qui modulent et contrôlent leurs interactions Les interactions physiques entre les molécules d'une cellule ou d'un compartiment sub-cellulaire traduisent, entre autre, l'aptitude structurale de ces molécules à se reconnaître.
Exemples :
|
c. Forces de liaison qui maintiennent la structure des macromolécules Ces forces sont non covalentes ou covalentes (ponts disulfures dans le cas des protéines) et très variées en nombre et du point de vue énergétique. Dans le cas des protéines, ces forces sont intimement liées aux propriétés physico-chimiques des résidus d'acides aminés donc liées aux conditions cellulaires (pH, température, viscosité, pression). Tous ces paramètres physico-chimiques sont maintenus relativement constants dans la cellule.
Ces paramètres physico-chimiques contrôlent ces équilibres, donc la flexibilité ou dynamique conformationnelle de toutes les molécules biologiques.
|
d. La notion d'affinité entre macromolécules biologiques C'est la caractéristique qui traduit la propension, dans un environnement et des conditions cellulaires donnés, de 2 (ou plus) macromolécules biologiques à se reconnaître et à interagir de manière réversible. Outre la complémentarité de structure, le paramètre clé de l'interaction entre molécules est leur concentration respective.
L'affinité de liaison est influencée par les paramètres physico-chimiques qui influencent la structure des macromolécules biologiques qui interagissent :
|
e. Rôle majeur des modifications post-traductionnelles dans les interactions protéines-protéines Les modifications post-traductionnelles (MPT) jouent un rôle déterminant dans la diversité des protéines (et leur localisation). Les MPT ont donc une importance capitale dans les interactions protéine-protéine et participent ainsi à la régulation de pratiquement tous les processus cellulaires.
Source : Virag et al. (2020) Le protéome humain est dynamique : il varie en réponse à une multitude de stimuli et les MPT régulent l'activité cellulaire. Selon la littérature, on estime que environ 5 % du protéome correspond aux enzymes qui catalysent 200 à 400 types de MPT :
Les MPT peuvent être introduites dans les protéines n'importe quand au cours de leur existence :
|
Stratégies pour identifier les interactions protéine-protéine médiées par des MPT Il n'est pas aisé de développer des méthodes fiables pour identifier des protéines interagissant exclusivement avec une MPT d'une autre protéine. En effet :
|
| Stratégies pour générer et analyser des MPT des protéines | ||
| Stratégie | Avantages | Limitations |
| Protéines modifiées dans les cellules dans des conditions contrôlées | Utilisation simple et peu de manipulations. L'absence de MPT peut être facilement modélisée en mutant le site cible ou en éliminant/désactivant la protéine qui crée la MPT. |
Les conditions pour introduire de nombreuses MPT ne sont pas complètement connues. Le degré de modification endogène est souvent hétérogène. La modification spécifique d'un site MPT peut être difficile. |
| Utilisation de peptides synthétiques contenant la MPT | Génération facile par synthèse chimique. Etude d'une grande variété de MPT. Possibilité d'introduire plusieurs MPT au sein du même peptide. Possibilité d'ajouter des photo-réticulateurs. Peut être utilisé dans un format à débit plus élevé tel que les puces de peptides. |
Ne traduit pas le comportement de la protéine entière repliée, ce qui peut abolir partiellement ou complètement l'interaction avec le(s) partenaire(s). Ne peut pas être utilisé pour étudier les interactions protéine-protéine induites par une MPT qui n'impliquent pas directement le résidu modifié. |
| Utilisation d'acides aminés naturels qui imitent une MPT | Facile à mettre en œuvre par mutagenèse simple. Introduit des MPT sur des sites spécifiques. |
Des différences importantes de structure entrainent un mimétisme imparfait. La plupart des MPT ne peuvent pas être simulées par un acide aminé naturel. |
| Utilisation de la ou des protéine(s) endogène(s) qui crée(nt) une MPT | Relativement simple avec des protéines de modification recombinantes. La modification de protéines qui introduisent des MPT permet de modifier des protéines substrat spécifiques. |
L'origine biochimique de nombreuses MPT est inconnue ou difficile à reconstituer. Il peut être difficile de modifier de manière homogène des sites spécifiques. Certaines MPT ne sont pas introduites par voie enzymatique. |
| Semi-synthèse protéique par ligature de protéines exprimées ("Expressed Protein Ligation" - EPL) | Synthèse de protéines pleine longueur modifiées de manière homogène. Synthèse d'une grande variété de MPT naturelles ou synthétiques. Plusieurs MPT différentes peuvent être introduites. |
Technique complexe et exigeante. L'EPL est réalisée dans des conditions dénaturantes : replier la protéine résultante peut être difficile. Expériences dans des cellules vivantes en général impossibles. Les MPT internes sur de longues protéines sont difficiles à analyser. |
| Expansion du code génétique | Des protéines de pleine longueur modifiées de manière homogène peuvent être générées. Des similitudes de structure proches des MPT peuvent être incorporées sans être supprimées. Les protéines modifiées peuvent être exprimées dans des cellules vivantes. Le résidu modifié peut théoriquement être incorporé dans n'importe quel site d'une protéine recombinante. |
Un nombre limité de MPT ont été génétiquement codées. L'efficacité de l'incorporation dépend du site d'incorporation. L'incorporation de multiples MPT dans une protéine peut être difficile. |
| Source : Wang et al. (2022) | ||
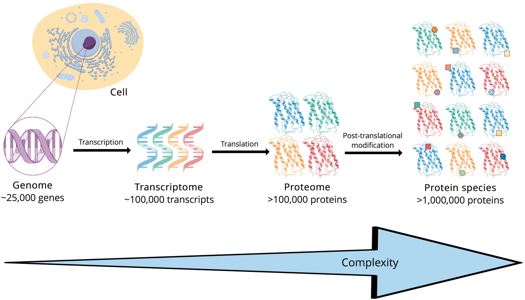
2. Identification et quantification des protéines a. Les protéomes et la protéomique La protéomique a pour but d'identifier (et de quantifier) l'ensemble des protéines synthétisées ou protéome, à un moment donné et dans des conditions données au sein d'un tissu, d'une cellule ou d'un compartiment cellulaire. Le protéome est extrêmement complexe à plusieurs titres :
|
| Grandes étapes du génome et du protéome de l'homme | ||
| Analyse aboutie du génome de l'homme | 2012 | "The ENCODE Project Consortium" :
20 687 gènes codant des protéines Nature 489, 57-74 (2012) |
| Protéome de l'homme | 2014 | Kim et al. (2014) "A draft map of the human proteome" Nature 509, 575-581 Bases de données : Human Proteome Map et ProteomicsDB |
| Analyse conjointe du génome et du protéome (5 niveaux d'évidence d'existence des protéines) de l'homme | 2018 - 2020 | 19 823 gènes codant des protéines 20 399 protéines (dont 17 694 protéines PE1) |
| Voir la liste des protéomes (en particulier les protéomes de référence) dans la base de données UNIPROT. | ||
|
La protéomique spatiale est en pleine expansion (techniques de plus en plus performantes et bases de données de plus en plus exhaustives) : elle fournit des informations essentielles sur l'organisation spatiale des protéines dans les tissus. La protéomique spatiale est à la base de nombreux projets d'atlas mondiaux. Exemple : "The human protein atlas". Le terme "protéomique spatiale" est générique : il englobe un très large éventail de méthodes basées sur l'immunohistochimie. En particulier :
Pus récente, la technique de protéomique visuelle profonde ("Deep Visual Proteomics" - DVP) :
Toutes ces méthodes (conjuguées ou non) génèrent des images hautement multiplexées d'échantillons telles que des tranches de tissus ou d'organes : elles dévoilent ainsi leur composition protéique et l'organisation spatiale de ces protéines. Par ailleurs, ces méthodes sont accompagnées d'algorithmes d'apprentissage profond pour l'analyse du très grand nombre et de la diversité de données qu'elles génèrent. De très nombreux laboratoires (entreprises privées ou publics) développent des outils d'analyse via le WEB appelés plateforme d'imagerie tissulaire multiplexée ("Multiplexed Tissue Imaging" - MTI). |
|
Voir un cours sur l'interactomique et les réseaux d'interactions protéine-protéine. a. Caractéristiques et paramètres clés des interactions entre molécules biologiques Les interactions qu'établissent les milliards de molécules au sein d'une cellule et avec l'extérieur sont l'élément clé de tous les processus biologiques, donc du fonctionnement cellulaire, donc de la vie. L’un des paramètres clés des interactions protéine-protéine est donc l'abondance des protéines qui interagissent.
Les techniques de multi-omiques spatiales appliquées aux cellules individualisées ("single-cell spatial multi-omics approaches") permettent d’établir un panorama complet d’une précision inégalée de la répartition, de l’abondance et du temps de demi-vie des protéines (en particulier), des ARN de divers types et du taux de transcription des gènes (en général). |
b. Les interactomes impliquant les protéines L'interactome des protéines correspond à l'ensemble des interactions protéine-protéine ("Protein - Protein Interactions" - PPI). Exemple d'interactome de l'homme : "The Human Reference Protein Interactome Mapping Project" - HuRI.
Source : Singh A. (2024)
Beaucoup de protéines sont nativement non structurées. Cette caractéristique accentue le caractère transitoire des interactions entre protéines (ou entre protéine et ligand au sens large). De plus, les molécules d'eau (d'hydratation des protéines ou intrinsèques à la stabilisation de la structure des protéines) jouent un rôle primordial dans la dynamique conformationnelle des protéines, donc dans leur interactivité. |
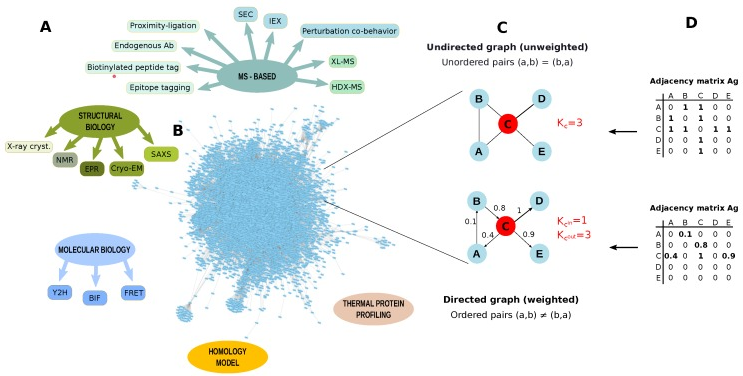
c. Démarche générale pour l'identification des interactions protéine-protéine
Source : Di Silvestre et al. (2018) Figure ci-dessus : (A) Exemples de méthodes biologiques, biophysiques ou associées à la spectromètrie de masse, pour identifier des PPI. |
4. Quantification de l'interaction entre macromolécules biologiques a. Constantes d'équilibre d'association KA et de dissociation KD Toute réaction d'association (inversement de dissociation) entre 2 (ou plus) molécules M1 et M2 peut s'écrire : M1 + M2 <=> M1-M2 Cette réaction d'association est régie par une constante d'équilibre d'association KA (inversement, par une constante d'équilibre de dissociation KD) quantifiables si on dispose d'une méthode permettant :
Exemple de l'équilibre de fixation d'un ligand L sur une protéine P : La vitesse d'association s'écrit : va = ka . [P].[L] - La vitesse de dissociation s'écrit : vd = kd . [PL]
Quand le système est à l'équilibre, les vitesses d'association et de dissociation sont égales :
Source : Xing et al. (2016) Plus KD est faible, plus l'affinité entre le ligand et la protéine est élevée. La liaison [biotine - streptavidine] et la liaison [inhibiteur de la ribonucléase - ribonucléase] sont caractérisées par KD ≈ 10-15 M (ou KA ≈ 1015 M-1) et sont parmi les interactions biologiques les plus fortes connues. |
|
Exemples d'association protéine - ligand
|
|
| hémoglobine - oxgène | le complexe enzyme - substrat(s) |
| antigène - anticorps | enzyme - régulateur (inhibiteur, activateur, coenzymes, ...) |
| histones - ADN (épigénétique) | protéine de transport - soluté spécifique |
| facteur de transcription - élément de réponse (gène) | récepteur - hormone (ou toute autre forme de signal) |
| protéine chaperon - protéine à replier | calmoduline - protéine cible activée |
| ribosome (complexe ribonucléoprotéique) - ARN messager (traduction) | l'eau : "ligand" universel des toutes les molécules biologiques |
b. Développement théorique des équilibres d'association - dissociation Equilibre de fixation d'un ligand sur une protéine et représentation de Scatchard. Ensemble d'excercices : détermination du nombre de site(s) de fixation d'un ligand et de la constante KD. Cours sur l'interactomique et les réseaux d'interactions protéine-protéine. |
c. Affinité, spécificité et multivalence des interactions entre protéines L'affinité est généralement quantifiée par la constante de dissociation à l'équilibre KD. Les interactions basées sur les motifs de type SLiM ont une spécificité particulière : des SLiM différents peuvent se lier au même domaine avec des affinités similaires et, inversement, plusieurs domaines peuvent se lier au même SLiM. La spécificité est une indication qualitative : elle traduit qu'une protéine donnée reconnaît mieux une certaine molécule (un ligand) que d'autres molécules (d’autres ligands). Si cette protéine reconnaît simultanément plusieurs ligands, on emploie la notion de sélectivité. Traduire les interactions entre protéines en terme de spécificité plutôt que d'affinité présente l’avantage de s’affranchir des paramètres physico-chimiques (pH, force ionique, encombrement stérique, ...) qui influencent plus fortement l'affinité. Différents modes de spécificité dans les interactions protéine-protéine impliquant des motifs de reconnaissance désordonnés
Source : Ivarsson & Jemth (2019)
PRISMA ("PRotein Interaction Screen on a peptide MAtrix") associée à la spectromètrie de masse quantitative permet d'identifier et cartographier les motifs d'interaction de type SLiM et MoRF ("Molecular Recognition Features" - séquences de 10 à 70 residus d'acides aminés) des protéines. |
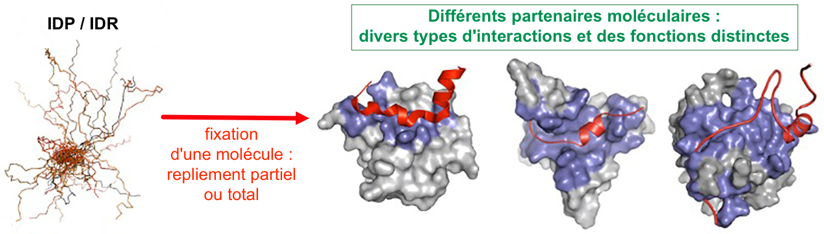
5. Les protéines ou régions intrinsèquement désordonnées (IDP/IDR) a. Introduction et définitions Certaines protéines sont fonctionnelles sans avoir une structure native pleinement ordonnée/structurée, aussi bien chez les procaryotes que chez les eucaryotes. Ces protéines ou régions intrinsèquement désordonnées ("Intrinsically Disordered Proteins or Regions" - IDP/IDR) :
De nombreuses interactions protéine-protéine résultent de la liaison, après leur repliement, des IDR de l’une des protéines, à un domaine replié de l’autre protéine ou (moins fréquemment) à une IDR de cette autre protéine. Les séquences de liaison désordonnées les plus courtes (3 à 12 résidus d’acides aminés) sont appelées motifs linéaires courts ("Short Linear Motifs" - SLiM) ou motifs linéaires eucaryotes ("Eukaryotic Linear Motifs" - ELM) et sont extrêmement fréquents dans les protéomes des eucaryotes :
Voir un cours sur les IDP/IDR et les acides aminés qui en sont spécifiques. |
b. Rôles biologiques des IDP/IDR et avantages pour la cellule En raison de leur plasticité conformationnelle, les IDP/IDR s'associent à des partenaires moléculaires avec lesquels des protéines complètement repliées ne pourraient interagir. Par ailleurs, certaines IDP/IDR sont dotées de la propriété de promiscuité : une même région d'une IDP/IDR fixe plusieurs partenaires et agit comme une "plaque tournante" dans les réseaux d'interactions protéine-protéine qui sont au coeur des processus de signalisation cellulaire. Comme toutes les interactions protéine-protéine, les interactions [IDP/IDR - partenaire moléculaire] sont modulées par l'environnement ou par des modifications covalentes. Quelques caractéristiques des IDP/IDR qui en démontrent le potentiel pour la cellule :
Figure adaptée de : Chakrabarti & Chakravarty (2022) |
c. Contraintes imposées par la chiralité des acides aminés dans les interactions La plupart des protéines naturelles sont lévogyres (L) car constituées d'acides aminés L uniquement. Les acides aminés dextrogyres (D) naturels sont donc rares. L'étude des contraintes imposées par la chiralité des acides aminés dans les interactions au sein de 5 couples de protéines en interaction indique :
Exemple de couple L'interaction de la prothymosine-α (ProTα) avec l'histone H1.0 (H1) s'est avérée une interaction désordonnée à haute affinité.
Les affinités de liaison ont été mesurées par ITC ("Isothermal titration calorimetry") et par spectroscopie FRET à molécule unique (smFRET) en marquant ProTα avec des fluorophores donneurs et accepteurs.
|
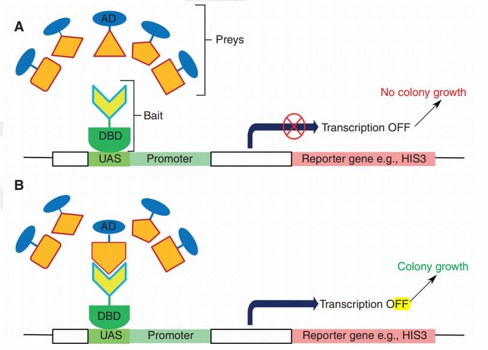
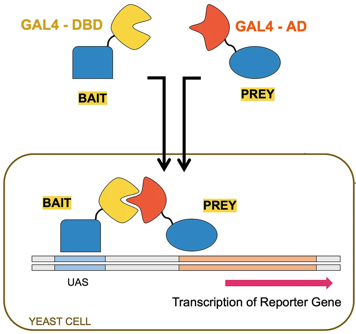
6. Quelques méthodes pour prouver les interactions protéine-protéine Voir un cours détaillé sur l'interactomique. a. La technique de complémentation double-hybride La construction génétique appelée double-hybride dans la levure ("Yeast Two-Hybrid system", "Two-hybrid screening" ou "Yeast two-Hybrid" - Y2H) est une technique très haut débit. Cette construction génétique utilise les propriétés structurales et fonctionnelles du facteur de transcription GAL4 ("Regulatory protein GAL4" - 881 acides aminés) de la levure Saccharomyces cerevisiae. |
Gal4 contient un domaine N-terminal appelé "DNA Binding Domain" - DBD (acides aminés 1 à 147) . Ce doamine se fixe à la séquence d'activation située en amont ("Upstream Activating Sequence" - UAS) d'un gène rapporteur ("reporter gene"). |
Gal4 contient un domaine C-terminal appelé "Activation Domain" - AD (acides aminés 768 à 881). Ce domaine est responsable de l'initiation de la transcription en aval de l'UAS quand il se fixe à d'autres composants de la machinerie de la transcription. |
| Ce domaine est fusionné à une protéine dite "appât" ("bait"). | Différentes constructions permettent de fusionner ce domaine à différentes protéines dites "proies" ("prey"). |
|
Source : Mehla et al. (2015) |
|
Principe de la technique de complémentation double-hybride
Source : Conde J.N. (2022) Avantages de cette technique
Limitations de cette technique
|
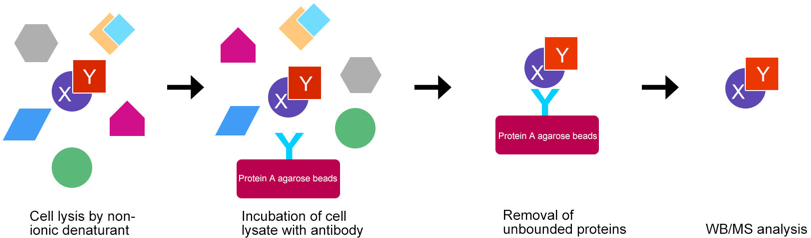
b. Autres méthodes de complémentation ou biochimiques
|
Techniques d'immunoprécipitation
Source : Biologics International Corp.
|
c. Méthodes physiques
d. Méthodes bioinformatiques Les méthodes issues de l'apprentissage profond. Notamment :
|
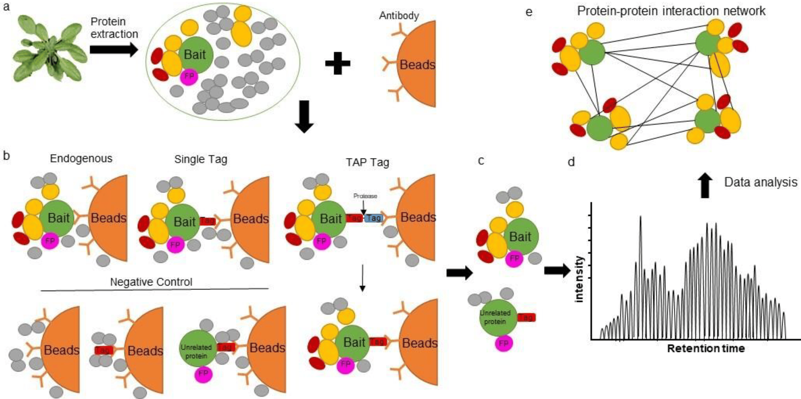
7. La purification par chromatographie d'affinité couplée à la spectromètrie de masse ("Affinity-Purification coupled to Mass Spectrometry") - AP-MS
Principe de l'AP-MS
Figure a ci-dessous
Figure b
Source : Kerbler et al. (2021) Figure c
Figure d : des séquences des protéines purifiées sont fractionnées, séparées et analysées par une technique de spectrométrie de masse. Figure e : l'analyse bioinformatique et biostatistique des données permet, entre autre, d'établir un réseau d'interactions protéine-protéine. |
| Exemples d'étiquettes | |||
| Etiquette | Séquence ou taille | Molécule greffée sur la résine d'affinité | Condition(s) d'élution de la résine d'affinité |
| TAPi | 45 kDa | Peptide de liaison à la calmoduline avec 2 domaines Protéine A | Protéine A - abaissement du pH |
| Peptide de liaison à la streptavidine (SBP) | WSHPQFEK | Streptavidine | Desthiobiotine |
| GSyellow | 37 kDa | Étiquette peptidique se liant à la streptavidine avec protéine fluorescente jaune citrine | Desthiobiotine et changement de pH |
| Protéines fluorescentes (GFP, YFP) | 26.9 kDa | Anti-GFP | pH |
| GSrhino | 21.9 kDa | 2 domaines de liaison aux IgG de la protéine G et 1 étiquette SBP | Streptavidine |
| TAPa | 26 kDa | Domaine de liaison IgG avec XHis et Xmyc | Protéolyse avec HR3C - imidazole - abaissement du pH |
| TAP ("Tandem Affinity Purification") : purification par affinité en tandem (2 étiquettes) SBP : "Streptavidin binding peptide" Étiquette GSrhino : 2 domaines de liaison à l'IgG anti-protéine G et 1 SBP séparés par 2 sites de protéolyse par la protéase HR3C ("Human Rhinovirus 3C") GFP : "Green Fluorescent Protein" ; YFP : "Yellow Fluorescent Protein" TAPa : "Alternative TAP" |
|||
La streptavidine est une protéine de la bactérie Streptomyces avidinii.
|
|||
| 8. Application de la fluorescence à l'analyse des macromolécules biologiques
a. Principe de la fluorescence L'énergie E d'un photon est proportionnelle à sa fréquence ν (h est la constante de Planck) : E = hν Pour qu'un électron passe de l'état fondamental à un état excité, il faut qu'il reçoive une quantité d'énergie équivalente à la différence d'énergie entre ces deux niveaux d'énergie.
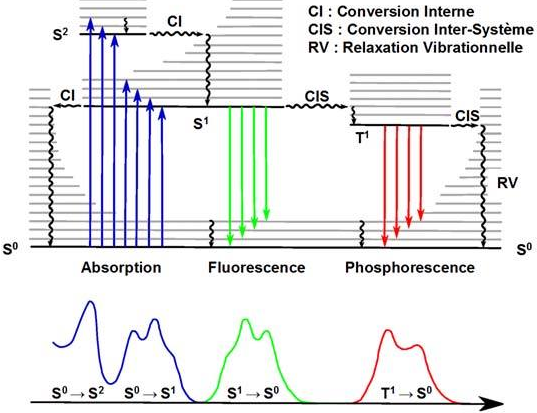
Un diagramme de Jablonski (ou Perrin - Jablonski) représente les transitions entre les différents états électroniques d'une molécule à l'origine des différents mécanismes : absorption, fluorescence, phosphorescence, mécanismes non-radiatifs, ... (voir A. Jablonski).
Source : Starck M. (2010) Dans le cas d'une molécule complexe comme une protéine, les niveaux énergétiques S0 et S1 sont multiples : il y a des pertes d'énergie au sein de la protéine lors du retour d'un sous-état excité S1' à un sous-état excité S1 (exemple ci-dessus).
Remarques :
|
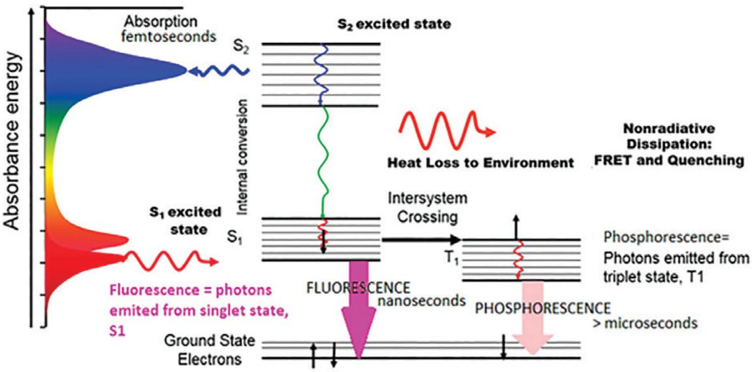
Processus non radiatifs Outre par émission de fluorescence, l'état excité S1 peut revenir à l'état fondamental S0 par deux mécanismes dits non radiatifs. Ces mécanismes sont en compétition avec l'émission de fluorescence et en diminuent le rendement (diminution de l'intensité de fluorescence ou "quenching").
Source : Horiba Scientific |
b. La diminution de l'intensité de fluorescence ou extinction de fluorescence ("fluorescence quenching") La réaction physico-chimique de ce phénomène est schématiquement : Mf*+D -> M+D La cinétique de désactivation suit l'équation de Stern - Volmer :
La représentation f0/f[Q] - 1 = f([Q]) est donc une droite de pente KSV.
Remarque : seule une fraction des collisions [fluorophore - désactivateur] est efficace pour la désactivation. La valeur réelle de kq ne peut donc être déterminée qu'expérimentalement. |
c. Le développement et la performance de molécules fluorescentes Du fait de son extrême sensibilité et de son aspect quantitatif (comptage des désintégrations), la radioactivité d'isotopes d'atomes constitutifs des molécules biologiques a été pendant de très longues années la méthode de choix pour décrypter les processus biologiques (cycle de Krebs, cycle de Calvin, méthode d'origine de séquençage de Sanger, …). Inconvénients de l'utilisation de radioéléments :
|
Définitions Une molécule fluorescente (fluorophore ou fluorochrome) :
|
Le développement de fluorophores / fluorochromes adaptés à l'étude des molécules biologiques L'avènement du séquençage des génomes par des techniques remplaçant la méthode classique de Sanger a très largement contribué au développement très rapide d'une palette sans cesse croissante de molécules fluorescentes extrêmement efficaces. Leur seuil de détection est désormais de l'odre de la molécule individualisée ("single molecule"). Ces molécules peuvent être fixées de manière covalentes aux molécules biologiques.
Ces molécules ont révolutionné les performances des techniques d'imagerie cellulaire, en particulier en microscopie :
|
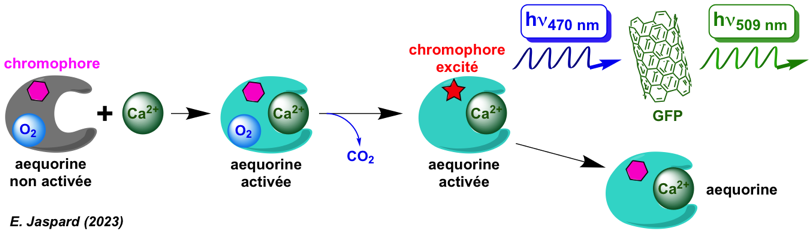
d. La protéine fluorescente verte (GFP) La protéine fluorescente verte ("Green Fluorescent Protein" - GFP) est synthétisée dans les photocytes (cellule qui produit de la bioluminescence) de la méduse Aequorea victoria. La découverte de cette protéine fluorescente et ses très nombreuses applications en biologie ont été couronnées par le prix Nobel de chimie 2008.
La GFP est un accepteur de transfert d'énergie :
|
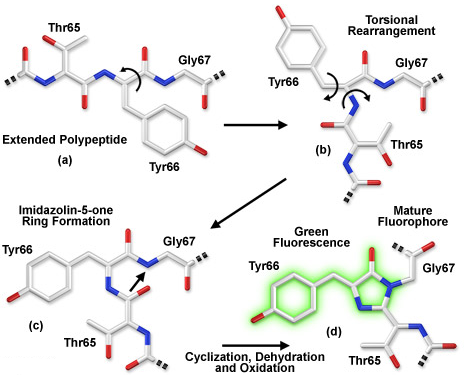
Le chromophore de la GFP
La fluorescence se produit lorsque l'oxydation de la liaison entre les carbones α et β de Y66 par l'oxygène moléculaire étend la conjugaison électronique du cycle imidazoline pour inclure le cycle phényl de Y et son substituant para-oxygène (figure d ci-dessous).
Source : Zeiss - Illustration du mécanisme de cyclisation du mutant S65T. |
Visualisation de la GFP de Aequorea victoria à une résolution de 1,90 Å Code PDB : 1EMA La structure cristalline de la GFP a révélé que le chromophore tripeptide cyclique est enfoui au centre d'un "tonneau β" à onze brins entrelacés. Au cours du repliement, le tripeptide est positionné au cœur du tonneau β : les réactions de cyclisation et de déshydratation nécessaires à la formation du chromophore mature ont alors lieu.
|
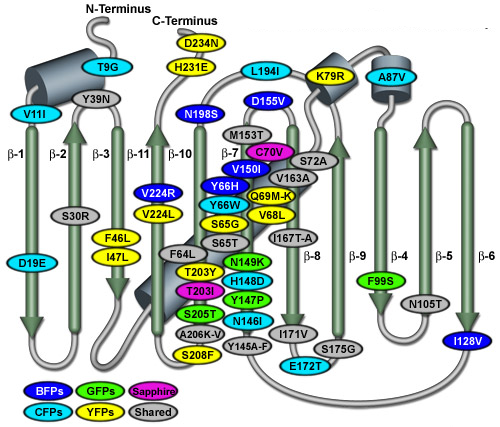
GFP et mutants de la GFP : utilisations très larges et intenses en biotechnologie La GFP a été mutée sur de nombreux résidus d'acides aminés afin de générer un très grand nombre de protéines fluorescentes avec des longueurs d'onde d'émission spécifiques. Voir la base de données de protéines fluorescentes FPbase. La figure suivante illustre diverses mutations de la GFP de Aequorea victoria (brins β = flèches vertes et hélices α = cylindres gris-bleus).
Source : Zeiss
|
| Protéine fluorescente | Séquence du chromophore | λexcitation (nm) | λémission (nm) |
| GFP ("Green Fluorescent Protein") | V61TTFSYGVQC70 | 395 | 509 |
| BFP ("Blue Fluorescent Protein") | V61TTFSHGVQC70 | 381 | 445 |
| CFP ("Cyan Fluorescent Protein") | V61TTFSWGVQC70 | 456 | 480 |
| YFP ("Yellow Fluorescent Protein") | L61VTTLGYGLM70 | 514 | 529 |
| Venus (YFP) | L61VTTLGYGLQ70 | 515 | 528 |
| Voir les paramètres physicochimiques d'une collection de ≈ 1000 protéines fluorescentes par ordre alphabétique (FPbase). | |||
Limitations des protéines de fusion [protéine fluorescente - protéine étudiée] La plupart des gènes disponibles dans le commerce pour la GFP et les protéines fluorescentes similaires contiennent environ 730 paires de bases.
Voir un développement concernant la GFP. Autres mutants de plus en plus performants Protéine fluorescente StayGold : dérivée par bioingéniérie de la protéine fluorescente verte (GFP) de la méduse Cytaeis uchidae.
Protéine fluorescente mBaoJin est un variant de StayGold sous forme de monomère. |
|
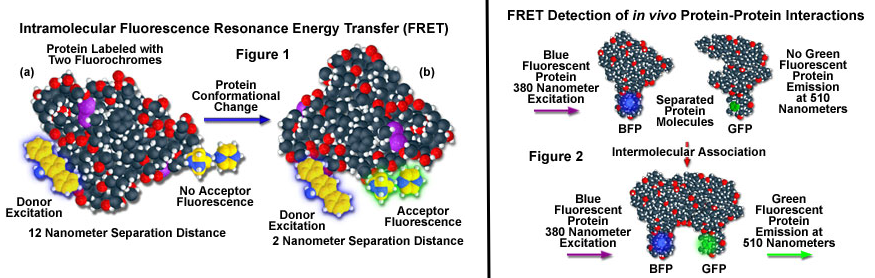
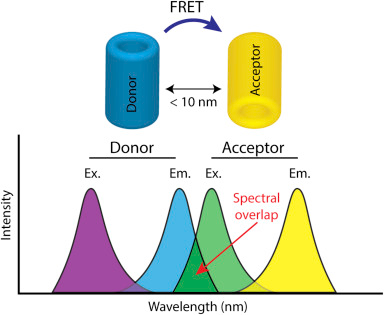
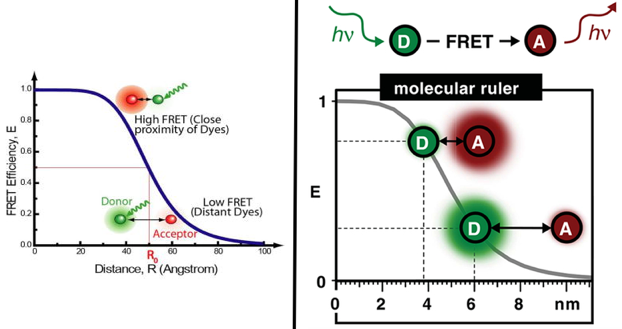
9. La technique du FRET appliquée aux molécules biologiques a. Principe du phénomène de FRET entre protéines fluorescentes Le FRET ("Förster Resonance Energy Transfer" - décrit par Theodor Förster en 1948) est un processus de transfert d'énergie entre une molécule fluorescente (un fluorophore appelé donneur) et une autre molécule fluorescente (un fluorophore appelé accepteur). Le phénomène FRET n'a lieu que si les 2 fluorophores sont distants de 10 Å à 100 Å. La technique du FRET, appliquée à la microscopie optique, permet donc d'analyser la proximité de deux biomolécules dans une cellule à l'échelle du nanomètre : elle permet de déterminer les interactions entre ces molécules quand elles se rapprochent suffisament l'une de l'autre.
Source : "Evident - Olympus" |
b. Principe physique du FRET Un électron de la molécule donneuse génère un champ électrique qui entre en résonnance avec les électrons des orbitales électroniques de la molécule acceptrice : la molécule acceptrice passe dans un état excité et son retour à l'état fondamental émet un photon (émission de fluorescence). Formule très théorique du calcul du FRET :
R0 = rayon de Förster, J(λ) = intégrale de chevauchement des aires, QD = rendement quantique, ε = coefficient d'extinction, κ = facteur d'orientation, n = indice de réfraction. Efficacité du FRET (E) C'est le rendement quantique du transfert d'énergie (la fraction de l'événement "transfert d'énergie" par événement "excitation du donneur de FRET") : E = kET / (kET + kf + ∑ki)
Dans le cadre d'une approximation ponctuelle [dipôle-dipôle], l'efficacité du FRET peut être reliée à la distance [donneur-accepteur] par les relations :
|
Chevauchement des spectres [émission de fluorescence du donneur - excitation (absorption) de fluorescence de l'accepteur] Pour augmenter l'efficacité du FRET, le groupe donneur doit avoir de bonnes capacités à émettre des photons : le groupe donneur doit être caractérisé par un coefficient d'extinction ε élevé et un rendement quantique élevé (voir ci-dessus). Le chevauchement du spectre d'émission du donneur et du spectre d'excitation (absorption) de l'accepteur traduit la quantité d'énergie émise par le donneur (préalablement excité) qui excite le groupe accepteur : plus les spectres se chevauchent, plus le donneur transfère de l'énergie à l'accepteur, plus le phénomène du FRET est important.
Source : Broussard & Green (2017) L'intégrale de chevauchement J(λ) [donneur - accepteur] représente l'amplitude du chevauchement des spectres. La valeur de cette intégrale est donnée par la relation :
FD(λ) est le spectre d'émission normalisé du donneur; εA(λ) est le coefficient d'absorption molaire de l'accepteur; λ est la longueur d'onde. |
c. Le couple [donneur / accepteur] du FRET La composition de ce couple est donc l'élément clé de la technique du FRET car le transfert d'énergie est d'autant plus important que le spectre d'émission de fluorescence du donneur et le spectre d'excitation (absorption) de l'accepteur se chevauchent. Exemples de couples [donneur / accepteur] fréquemment utilisés en biologie pour détecter des interactions protéine-protéine (entre macromolécules ou intramoléculaires) :
|
| Protéines fluorescentes utilisées dans diverses méthodes | |||
| Protéine (noms divers) | pic excitation (nm) | pic émission (nm) | Organisme |
| GFP ("Green Fluorescent Protein") | 513 | 527 | Aequorea victoria |
| CFP ("Cyan Fluorescent Protein" : ECFP, Cerulean, CyPet, mTurquoise2) mutation Y66W de la GFP |
433 | 475 | |
| YFP ("Yellow Fluorescent Protein" : Citrine, Venus, YPet) mutation T203Y de la GFP |
516 | 529 | |
| BFP ("Blue fluorescent protein" : EBFP, EBFP2, Azurite) mutation Y66H de la GFP |
383 | 448 | |
Illustration de BFP mKalama1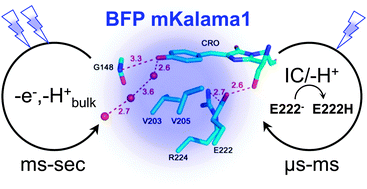 Source : Vegh et al. (2015) |
|||
La désintégration non radiative de l'état excité de BFP mKalama comprend deux évènements :
|
|||
| mCherry (mRFPs) | 587 | 610 | Discosoma sp |
FPbase : base de données de protéines fluorescentes ("Fluorescent Proteins" - FP) et de leurs propriétés.
|
|||
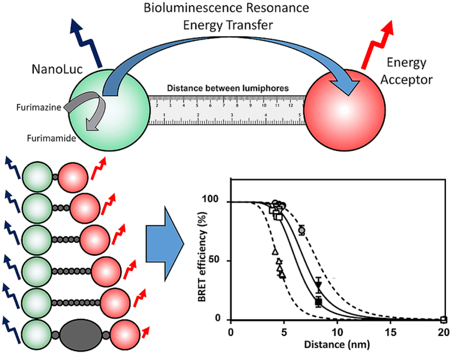
d. Le BRET ("Bioluminescence Resonance Energy Transfer") Cette technique mesure le transfert d'énergie entre une molécule bioluminescente donneur de BRET et une molécule fluorescente accepteur (par exemple la GFP ou la YFP, comme pour le FRET). Cette technique génère un bruit de fond très faible. La coelenterazine est une molécule bioluminescente donneur. C'est une luciférine dont l'excitation est déclenchée par la luciférase "renilla-luciferin 2-monooxygenase" (Rluc - E.C. 1.13.12.5). coelenterazine h + O2 <=> CO2 + coelenteramide h monoanion (excité) + H+ + hν Une luciférase extrêmement performante a été développée : la NanoLuc (NLuc).
Source : Weihs et al. (2020) |
10. Le FRET appliquée à l'étude de molécule unique ("single-molecule FRET") La dynamique des changements conformationnels d'une population de biomolécules n'est pas un processus homogène. En effet, les mouvements de ces molécules ne sont pas synchronisés et ils se déroulent sur une échelle de temps qui s'échelonne de la femtoseconde (fs; vibration des liaisons des chaînes latérales des rotamères) à la microseconde (µs; mouvement des structures secondaires) et, pour des mouvements de plus grande ampleur la seconde (mouvements globaux des domaines structuraux). Ces caractéristiques structurales sont donc extrêmement difficiles à analyser avec des méthodes d'imagerie reflétant le comportement moyen d'un vaste ensemble de molécules. La technique FRET à molécule unique ("single-molecule FRET" - smFRET) permet :
La technique smFRET est une application du phénomène de FRET dans laquelle des paires uniques de FRET [donneur et accepteur] sont excitées et détectées.
|
b. Le FRET dépend de la distance entre les deux fluorophores Le FRET est favorisé quand la distance [donneur – accepteur] est comprise entre 1 et 10 nm, ce qui souligne l'intérêt de smFRET. En effet, cette distance nécessaire au transfert d'énergie est du même ordre de grandeur que la taille d'une protéine ou l'épaisseur d'une membrane lipidique.
Sources : Roy et al. (2008) & Lerner et al. (2018) Rappel de l'éfficacité du transfert d'énergie en fonction de la distance [donneur-accepteur] :
La gamme de distances mesurées précisément par FRET [0,5 R0 - 1,5 R0] : cela se traduit par une plage dynamique de 2 à 10 nm pour les paires de colorants smFRET couramment utilisées. Exemple : R0 de la paire [Cy3 - Cy5] = 5,4 nm. Le fluorophore le mieux adapté aux études smFRET est :
|
Une excellente paire smFRET est caractérisée par :
Des protéines fluorescentes et des semi-conducteurs ont été utilisées pour des études smFRET sans réel succès. Les fluorophores monomoléculaires les plus efficaces s'avèrent les petits (< 1 nm) colorants organiques comme l'indique le tableau ci-dessous. |
| Propriétés spectrales de quelques fluorophores pour l'analyse de molécule individualisée ("single-molecule"). | ----- | Fluorophores | λmax excitation (nm) | λmax émission (nm) | εM à λmax (M-1.cm-1) | Rendement quantique | R0 (Å) |
| donneur | Cy3 | 555 | 570 | 150 000 | 0,31 | 54 |
| accepteur | Cy5 | 646 | 662 | 250 000 | 0,20 | |
| donneur | sulfo-Cy3 | 548 | 563 | 162 000 | 0,10 | 56 |
| accepteur | sulfo-Cy5 | 646 | 662 | 271 000 | 0,28 | |
| donneur | Cy3B | 558 | 572 | 130 000 | 0,67 | 51 |
| accepteur | Cy7 | 750 | 773 | 199 000 | 0,30 | |
| donneur | Alexa Fluor 555 | 555 | 580 | 155 000 | 0,10 | 47 |
| accepteur | Alexa Fluor 647 | 650 | 665 | 270 000 | 0,33 | |
| donneur | Atto 550 | 554 | 574 | 120 000 | 0,80 | 63 |
| accepteur | Atto 647 N | 644 | 667 | 150 000 | 0,65 | |
| donneur | Alexa Fluor 488 | 490 | 525 | 73 000 | 0,92 | 62 |
| accepteur | Alexa Fluor 568 | 578 | 603 | 88 000 | 0,69 | |
Figure ci-dessous, exemple du pigment "ATTO 550".
Sources : Atto-Tech Voir une application des cyanines en protéomique ("technique 2D-DIGE). |
c. Application du smFRET à l'étude de processus biologiques smFRET a été utilisé pour étudier divers types de processus et de systémes biomoléculaires.
Illustration des machineries moléculaires ClpB est une machinerie protéolytique homohexamérique de bactéries appartenant à la famille des ATPases Hsp100 associées à diverses activités cellulaires ("ATPases associated with diverse cellular activities" - AAA+). Chaque sous-unité de ClpB contient un domaine structural intermédiaire unique en spirale (le domaine M) qui est un élément de contrôle de la fixation du co-chaperon DnaK. La technique sMFRET a permis de démontrer que le domaine M bascule entre 2 états conformationnels majeurs sur une échelle de temps très rapide d'environ 150 µs (plus rapides que l'activité globale de ClpB). |
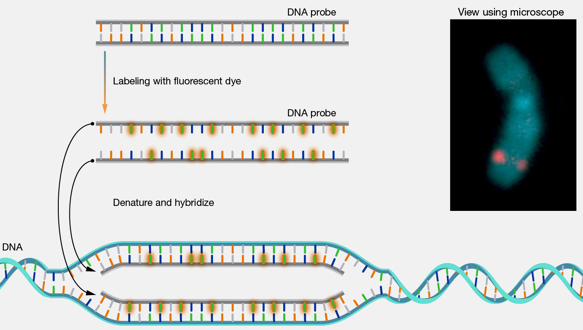
11. La technique FISH ("Enhanced ELectric Fluorescence in situ Hybridization") a. Principe L'hybridation in situ par fluorescence ou FISH est une technique cytogénétique moléculaire qui détecte et localise une séquence d'ADN spécifique sur les chromosomes entiers.
Source : National Human Genome Research Institute La technique FISH est employée pour le diagnostic de maladies génétiques, pour la cartographie génétique et l'identification d'anomalies chromosomiques et pour la comparaison d'arrangements chromosomiques de gènes d'espèces apparentées. |
b. Application du FISH à la transcriptomique spatiale à haut débit Les méthodes de détermination du profil transcriptomique dans l'espace tissulaire sont assujetties à un équilibre entre entre résolution et débit. La méthode nommée EEL-FISH ("Enhanced ELectric Fluorescence in situ Hybridization") traite rapidement de très nombreux échantillons de tissus sans perte de résolution spatiale. Les ARN d'une section de tissu sont transférés par électrophorèse sur une surface qui les capture.
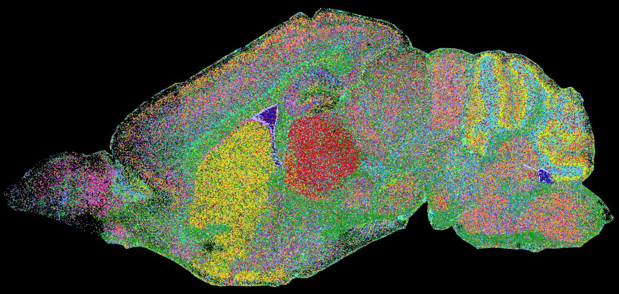
La méthode EEL-FISH appliquée à 8 sections entières d'un cerveau de souris a permis de mesurer le taux de transcription de 440 gènes, révéler ainsi son organisation tissulaire complexe (voir la base de données "Mouse Brain Atlas").
Source : Borm et al. (2023) Cette méthode peut être utilisée pour l'étude d'échantillons humains (difficiles à analyser) en supprimant la lipofuscine autofluorescente : le transcriptome spatial du cortex visuel humain peut-être ainsi visualisé. La lipofuscine est le nom donné aux fins granules pigmentaires jaune-brun composés de résidus lipidiques issus de la digestion par les lysosomes. |
12. Apprentissage profond et prédiction des interactions protéine-protéine Comme dans tous les domaines de la biologie (et d'autres disciplines), les méthodes issues de l'apprentissage machine apportent des informations complémentaires en traitant une quantité de données qu'aucune autre méthode ne peut traiter. Ces informations, aussi riches soient-elles, ne sont que prédictives (théoriques) et leur obtention s'appuient sur des modèles plus ou moins sophistiqués et représentatifs de la réalité biologique. Il est donc capital de ne pas perdre de vue que seule la confirmation expérimentale est une preuve tangible. Un nombre croissant de méthodes sont développées : l'une des difficultés est de faire un choix pertinent compte-tenu de la problèmatique biologique et des connaissances en informatique et en modèles mathématiques nécessaires pour effectuer ce choix. Voir un développement de l'intelligence artificielle en biologie. |
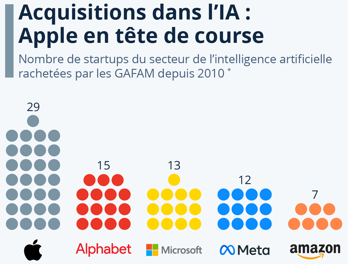
a. Prédominance des GAFAM dans les domaines de l'intelligence artificielle Les GAFAM sont les pionniers et dorénavant les leaders du marché de l'apprentissage machine.
Source : Statista |
| Nom | But | Développeur |
| TensorFlow (écrit en C++ et en Python) |
Bibliothèque logicielle pour l'apprentissage automatique, en particulier la formation et l'inférence de réseaux neuronaux. | Développé par Alphabet (Google) |
| PyTorch (écrit en C++, Python et autres) |
Bibliothèque d'apprentissage automatique basée sur la bibliothèque Torch. | Initialement développée par Meta-AI et désormais gérée par Linux Foundation |
| Scikit-learn (écrit en Python) |
Bibliothèque d'apprentissage machine construite sur la base des bibliothèques NumPy, SciPy et Matplotlib. | Le projet d'origine (2007) est issu du programme annuel organisé par Google ("Google Summer of Code"). |
| Keras (écrit en Python) |
Bibliothèque qui permet d'interagir avec les algorithmes de réseaux de neurones profonds et d'apprentissage automatique tels que Tensorflow ou PyTorch. | Développé dans le cadre du projet ONEIROS en grande partie par un ingénieur de Google (F. Chollet). |
| ChatGPT (écrit en Python) |
Agent conversationnel |
Développé par OpenAI dont l'un des principaux investisseur est Microsoft. |
| Le langage Python est très largement employé pour écrire les programmes d'apprentissage machine (et bien d'autres programmes) en biologie. | ||
| Exemples de logiciels et de modèles basés sur l'apprentissage machine pour la prédiction d'interactions protéine-protéine | ||
| Nom | Algorithme | But |
| Prédiction de la structure de la protéine cible | ||
| Serveur trRosetta AlphaFold |
DNN | Prédiction de la structure 3D des protéines |
| Complexe QA | GNN | Prédiction de la structure 3D des complexes protéiques |
| Protein BERT | Transformeur | Prédiction des structures secondaires des protéines |
| ESMfold | Transformeur | Prédiction de la structure 3D des protéines |
| Prédiction des interactions protéine ? protéine | ||
| IntPred | RF | Prédiction de l'interface des sites d'interactions protéine-protéine |
| eFindSite | SVM - NBC | Prédiction de l'interface des d'interactions protéine-protéine |
| DELPHI | RNN - CNN | Prédiction des sites d'interactions protéine-protéine |
| PPISP-XGBoost | XGBoost | Prédiction des sites d'interactions protéine-protéine |
| HN-PPISP DeepSG2PPI |
CNN | Prédiction des sites d'interactions protéine-protéine |
| TAGPPI SGPPI DL-PPI |
GCN | Prédiction des interactions protéine-protéine |
| Struct2Graph | GAT | Prédiction des interactions protéine-protéine |
| DeepFE-PPI DeepPPI |
DNN | Prédiction des interactions protéine-protéine |
| MaTPIP | Transformeur - CNN | Prédiction des interactions protéine-protéine |
| ProtInteract | Autoencodeur - CNN | Prédiction des interactions protéine-protéine |
|
DNN - "deep neural network" : réseau de neurones profond ; GNN - "graph neural networks" : réseaux de neurones graphiques ; RNN - "recurrent neural network" : réseau de neurones récurrent ; RF - "random forest" : forêt aléatoire ; CNN - "convolutional neural network" : réseau de neurones convolutif ; GCN - "graph convolutional network" : réseau convolutif graphique ; GAT - "graph attention network" : réseau graphique avec attention ; SVM - "support vector machine" : machine à vecteurs de support ; NBC : "naïve Bayes classifier" : classificateur bayésien naïf ; XGBoost - "extreme gradient boosting" : amplification de gradient extrême.
Les méthodes RF et SVM réduisent la haute dimensionnalité des structures protéiques (remplacement des données dans un espace de grande dimension par celles dans un espace de plus petite dimension). |
||
| Source : Qi et al (2024) | ||
|
D-SCRIPT ("Deep Sequence Contact Residue Interaction Prediction Transfer") est une méthode de prédiction des interactions protéine-protéine basée sur la séquence, qui modélise la structure des protéines en utilisant un modèle de langage pré-entra?né.
Exemples de modèles de langage pré-entraînés : |
||
b. Apport des méthodes issues de l'apprentissage profond dans l'identification d'interactions protéine-protéine médiées par des modifications post-traductionnelles Un ensemble d'interactions protéine-protéine modulées par les modifications post-traductionnelles (MPT) chez l'homme a été extrait de la base de données IntAct : sur plus de 100.000 interactions protéine-protéine décrites, environ 3000 sont médiées par des MPT.
Enfin, ce dernier a été appliqué à la fouille textuelle de 18 millions de résumés d'articles de PubMed (2019) : environ 547.000 interactions protéine-protéine médiées par les MPT sont ainsi prédites dont environ 4600 avec une confiance élevée et une faible variation (Elangovan et al., 2022). |
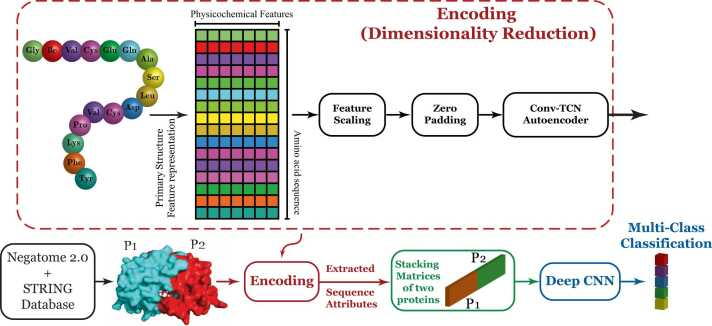
c. Illustration : le programme de prédiction ProtInteract Le programme ProtInteract est une méthode basée sur les séquences des protéines qui prédit les interactions protéine-protéine via deux tâches principales : l'encodage et la prédiction. Une architecture spécifique d'autoencodeur encode la séquence en acide aminés de chaque protéine :
Source : Soleymani et al. (2023) |
Le jeu d'entraînement de ProtInteract ProtInteract est entraîné avec les données d'interactions protéine-protéine issues de la base de données STRING :
Ces scores d'interaction sont ensuite normalisés entre 0 (paires de protéines qui n'interagissent pas) et 1 (paires de protéines qui interagissent avec la plus grande confiance). |
Encodage avec l'autoencodeur de ProtInteract ProtInteract encode la position des acides aminés de la séquence d'une protéine et certaines de leurs propriétés physicochimiques dans un espace vectoriel de dimension inférieure ("dimensionality reduction"). Chaque acide aminé est d'abord encodé ("encoding") selon 10 caractéristiques physicochimiques très informatives :
Cette étape extrait des propriétés hautement informatives de chaque séquence tout en réduisant la complexité des calculs effectués ultérieurement par les réseaux de neurones. |
Prédiction des interactions protéine-protéine de ProtInteract ProtInteract utilise ensuite ces informations pour prédire les interactions entre 2 protéines en fonction de leurs séquences en acides aminés. Cette prédiction correspond à 3 scénarios de classification des interaction protéine-protéine. Ces scénarios comprennent respectivement 2, 3 et 5 classes d'interaction protéine-protéine ("Multi-Class Classification").
Finalement, à partir de chaque scénario, un réseau de neurones convolutif profond ("deep Convolutional Neural Network" - deep CNN) évalue l'exactitude du classement prédit d'une interaction entre 2 protéines. |
| 13. Liens Internet et références bibliographiques | |
Cours en ligne "Protein-protein interactions" Pathway Figure OCR : extraction d'informations publiées dans la littérature. Pathway Commons The Human Reference Protein Interactome Mapping Project |
|
Base de données de protéines fluorescentes FPbase : collection de ≈ 1000 protéines fluorescentes (paramètres physicochimiques et ordre alphabétique) The Eukaryotic Linear Motif (ELM) resource MaxQuant : progiciel de protéomique quantitative pour l'analyse de grands jeux de données obtenues par spectrométrie de masse |
|
Guide de sélection de fluorophores "Proteins Double as Qubits, A Step That Could One Day Bridge Quantum Computing And Biology" META-SiM Projector |
|
Smith & Johnson (1988) "Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase" Gene 67, 31 - 40 Fields & Song (1989) "A novel genetic system to detect protein-protein interactions" Nature 340, 245 - 246 Guan & Dixon (1991) "Eukaryotic proteins expressed in Escherichia coli: an improved thrombin cleavage and purification procedure of fusion proteins with glutathione S-transferase" Anal. Biochem. 192, 262 - 267 Prasher et al. (1992) "Primary structure of the Aequorea victoria green-fluorescent protein" Gene 111, 229 - 233 |
|
Zhu et al. (2001) "Global analysis of protein activities using proteome chips" Science 293, 2101 – 2105 Chatr-aryamontri et al. (2007) "MINT: the Molecular INTeraction database" Nucleic Acids Res. 35, D572 - D574 Starck M. (2010) "Synthèse, propriétés photophysiques et marquage biologique par des complexes de lanthanides luminescents" Thèse de doctorat |
|
Pavlopoulos et al. (2011) "Using graph theory to analyze biological networks" BioData Min. 4, 10 Szklarczyk et al. (2011) "The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored" Nucleic Acids Res. 39, D561 - D568 Kerrien et al. (2012) "The IntAct molecular interaction database in 2012" Nucleic Acids Res. 04, D841- D846 |
|
|
Mehla et al. (2015) "The yeast two-hybrid system: a tool for mapping protein-protein interactions" Cold Spring Harb. Protoc. (5), 425 - 430 Xing et al. (2016) "Techniques for the analysis of protein-protein interactions in vivo" Plant Physiol. 171, 727 - 758 Fernandes et al. (2016) "Systematic analysis of the gerontome reveals links between aging and age-related diseases" Hum. Mol. Genet. 25, 4804 - 4818 |
|
|
Broussard & Green (2017) "Research Techniques Made Simple: Methodology and Applications of Förster Resonance Energy Transfer (FRET) Microscopy" J. Invest. Dermatol. 137, e185 - e191 Hu et al. (2017) "Functional Analysis of Human Hub Proteins and Their Interactors Involved in the Intrinsic Disorder-Enriched Interactions" Int. J. Mol. Sci. 18, 2761 Feig et al. (2017) "Crowding in Cellular Environments at an Atomistic Level from Computer Simulations" J. Phys. Chem. B 121, 34, 8009 - 8025 Di Silvestre et al. (2018) "Large Scale Proteomic Data and Network-Based Systems Biology Approaches to Explore the Plant World" Proteomes 6, 27 |
|
|
Lambert T.J. (2019) "FPbase: a community-editable fluorescent protein database" Nat. Methods 16, 277 - 278 Ivarsson & Jemth (2019) "Affinity and specificity of motif-based protein-protein interactions" Curr. Opin. Struct. Biol. 54, 26 - 33 Kumar et al. (2020) "ELM-the eukaryotic linear motif resource in 2020" Nucleic Acids Res. 48, D296-D306 Gordon et al. (2020) "A SARS-CoV-2 protein interaction map reveals targets for drug repurposing" Nature 583, 459 - 468 Gogl et al. (2020) "Dual Specificity PDZ- and 14-3-3-Binding Motifs: A Structural and Interactomics Study" Structure 28, 747 - 759 Weihs et al. (2020) "Experimental determination of the bioluminescence resonance energy transfer (BRET) Förster distances of NanoBRET and red-shifted BRET pairs" Anal. Chim. Acta X. 6, 100059 |
|
|
Kerbler et al. (2021) "From Affinity to Proximity Techniques to Investigate Protein Complexes in Plants" Int. J. Mol. Sci. 22, 7101 Lelek et al. (2021) "Single-molecule localization microscopy" Nat. Rev. Methods Primers 1, 39 |
|
|
Karatzas et al. (2022) "The network makeup artist (NORMA-2.0): distinguishing annotated groups in a network using innovative layout strategies" Bioinform. Adv. 2, vbac036 Chakrabarti & Chakravarty (2022) "Intrinsically disordered proteins/regions and insight into their biomolecular interactions" Biophys. Chem. 283, 106769 Soleymani et al. (2022) "Protein - protein interaction prediction with deep learning: A comprehensive review" Comput. Struct. Biotechnol. J. 20, 5316 - 5341 Hu et al. (2022) "Deep learning frameworks for protein - protein interaction prediction" Comput. Struct. Biotechnol. J. 20, 3223 - 3233 Hirano et al. (2022) "A highly photostable and bright green fluorescent protein" Nat. Biotechnol. 40, 1132 - 1142 Conde J.N. (2022) "Yeast two-hybrid system for mapping novel dengue protein interactions" Protocol 119 - 132 |
|
|
smFRET Roy et al. (2008) "A Practical Guide to Single Molecule FRET" Nat. Methods.5, 507 - 516 Sasmal et al. (2016) "Single-Molecule Fluorescence Resonance Energy Transfer in Molecular Biology" Nanoscale 8, 19928 – 19944 Lerner et al. (2018) "Toward dynamic structural biology: Two decades of single-molecule Förster resonance energy transfer" Science 359, eaan1133 Mazal et al. (2019) "Tunable microsecond dynamics of an allosteric switch regulate the activity of a AAA+ disaggregation machine" Nat. Commun. 10, 1438 Groves et al. (2023) "Single-molecule FRET for virology: 20 years of insight into protein structure and dynamics" Q. Rev. Biophys. 56, e3 |
|
Kim et al. (2023) "A proteome-scale map of the SARS-CoV-2-human contactome" Nat. Biotechnol. 41, 140 - 149 Kurbatov et al. (2023) "The Knowns and Unknowns in Protein–Metabolite Interactions" Int. J. Mol. Sci. 24, 4155 Liu et al. (2023) "Depicting a cellular space occupied by condensates" Mol Biol Cell. 34, tp2 Goodsell & Lasker (2023) "Integrative visualization of the molecular structure of a cellular microdomain" Protein Sci. 32, e4577 |
|
|
Borm et al. (2023) "Scalable in situ single-cell profiling by electrophoretic capture of mRNA using EEL FISH" Nat. Biotechnol. 41, 222 - 231 Reinhardt et al. (2023) "Ångström-resolution fluorescence microscopy" Nature 617, 711 - 716 Soleymani et al. (2023) "ProtInteract: A deep learning framework for predicting protein - protein interactions" Comput. Struct. Biotechnol. J. 21, 1324 - 1348 Pusara et al. (2023) "Accurate calculation of second osmotic virial coefficients of proteins using mixed Poisson–Boltzmann and extended DLVO theory" Mol. Syst. Des. Eng. 8, 1203 - 1219 |
|
|
Singh A. (2024) "Understanding protein interaction dynamics" Nat. Met. 21, 2226 - 2227 Qi et al. (2024) "Machine Learning Empowering Drug Discovery: Applications, Opportunities and Challenges" Molecules 29, 903 Grassmann et al. (2024) "Computational Approaches to Predict Protein−Protein Interactions in Crowded Cellular Environments" Chem. Rev. 124, 3932 - 3977 Feder et al. (2025) "A fluorescent-protein spin qubit" Nature 645, 73 - 79 |
|