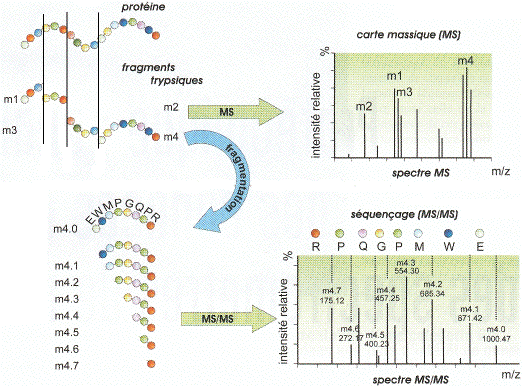
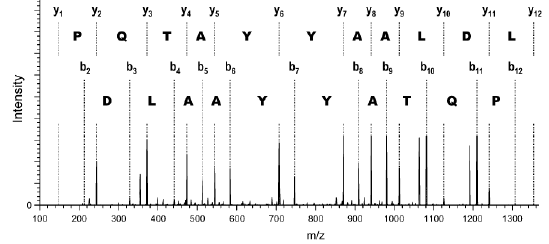
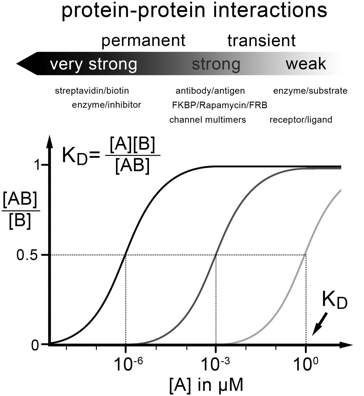
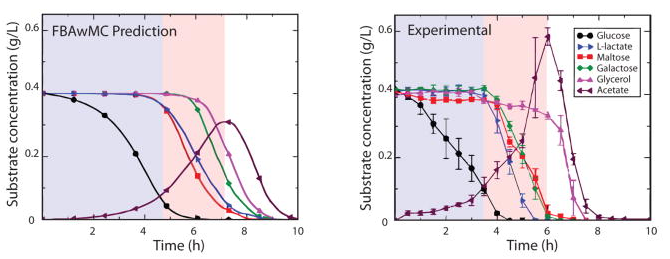
|
c. Exemples de technologies de séquençage dites de 3ème génération
α. Séquençage avec des nanopores (Minion & PromethION - Oxford Nanopore Technologies)
L'ADN est initialement hydrolysé en longs fragments de 8 à 10 kilo paires de bases.
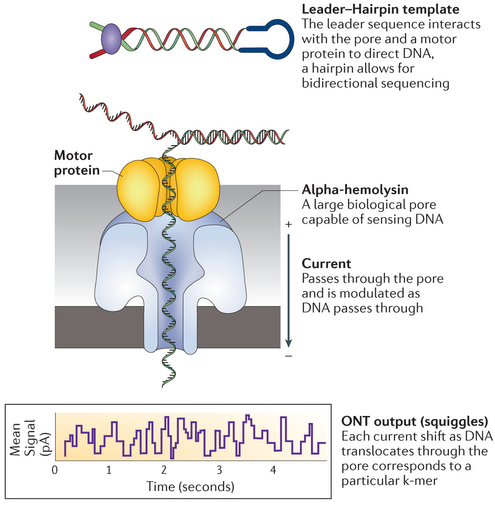
Deux adaptateurs différents ("leader" et "hairpin") sont attachés à l'une ou l'autre des extrémités de l'ADN. L'adaptateur "leader" est double-brin. Il contient :
- Une séquence qui dirige l'ADN au travers du pore.
- Une séquence d'attache qui dirige l'ADN vers la surface de la membrane.
Sans l'adaptateur "leader", les interactions de l'ADN avec le pore dans lequel l'ADN passe (voir ci-dessous) sont minimes, ce qui empêche tout fragment dont la conformation est "hairpin-hairpin" d'être séquencé.
- Actuellement, il n'existe aucune méthode pour diriger les adaptateurs vers une extrémité particulière.
- Il y a donc 3 conformations possibles de banque d'ADN : "leader-leader", "leader-hairpin" et "hairpin-hairpin" : la conformation idéale de la banque est donc "leader-hairpin".
Dans la conformation "leader-hairpin", la séquence "leader" dirige le fragment d'ADN à séquencer au travers d'un pore.
- Ce pore est l'α-hémolysine (protéine bactérienne heptamèrique de diamètre interne = 1 nm).
- Dans la nature, cette protéine s'insère dans la membrane des érythrocytes en formant un pore ce qui provoque, en général, la lyse de la cellule.
- Dans le système de séquençage, un courant électrique est généré quand les nucléotides du fragment d'ADN à séquencer traversent l'α-hémolysine.
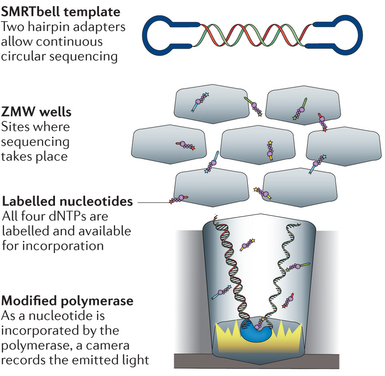
Source : Goodwin et al. (2016)
- A mesure que l'ADN traverse le pore, on observe un décalage caractéristique de l'intensité électrique (de l'ordre du pA).
- Divers paramètres (dont l'amplitude et la durée du décalage) sont enregistrés : ils correspondent à une séquence k-mer (k nucléotides successifs) particulière.
- Lorsque le nucléotide suivant traverse le pore, l'intensité électrique est différemment modulée et un nouveau k-mer est identifié.
Quand arrive l'extrémité "hairpin", c'est le brin complémentaire de l'ADN qui continue à être transloqué au travers l'adaptateur du pore : ainsi, les deux brins sont séquencés pour obtenir une séquence consensus dite "lecture 2D".
Les dernières avancées
Les technologies avec des nanopores sont de plus en plus performantes. En 2018, le séquençage et l'assemblage de novo d'un génome humain se sont appuyés sur un protocole :
- Qui a généré des lectures ultra-longues : N50 > 100 kb avec des longueurs de lecture jusqu'à 882 kb.
- La précision de l'assemblage (après incorporation des données de séquençage à lecture courte complémentaires) a dépassé 99,8%.
- Des lectures ultra-longues ont permis l'assemblage du locus du complexe majeur d'histocompatibilité de 4 Mo dans son intégralité.
- Voir Jain et al. (2018)
La technologie Minion est utilisée de longue date dans la station spatiale internationale ("International Space Station", ISS). Elle a été testée avec succès dans des conditions de gravité comparables à celles qui règnent sur Mars (G = 0,378), sur la lune (G = 0,166) et sur Europa (satellite de Jupiter - G = 0,134). Voir : Carr et al. (2020).
Le séquençage en routine du génome humain est devenu possible avec le séquenceur PromethION (Oxford Nanopore Technologies) qui possède 3000 capteurs et 12.000 pores : ils génèrent en moyenne 70 Go de données permettant une couverture 20X du génome humain.
Enfin, la précision des logiciel d'appel de base est sensiblement améliorée par des algorithmes basés sur des modèles de Markov cachés ou des réseaux de neurones. |