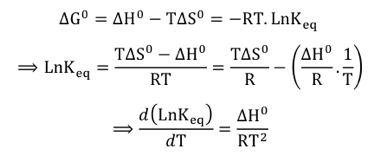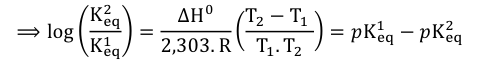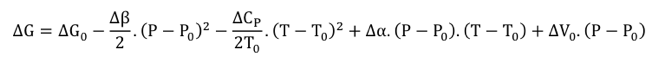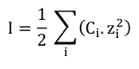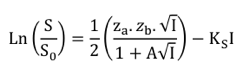| Effets de paramètres physico-chimiques (pH, température, pression, force ionique et viscosité) sur l'activité enzymatique |
| Tweet |
|
|
1. Effets du pH
2. Effets de la température
|
3. Effets de la pression
4. Effets de la force ionique
5. Effet de la viscosité du milieu 6. Liens Internet et références bibliographiques |
| Introduction
Les paramètres physico-chimiques tels que le pH, la température, la force ionique, la pression et d'autres, conditionnent l'activité des enzymes. Ce sont des caractéristiques particulières car la très grande majorité des organismes vit dans des conditions qui correspondent à une plage réduite de ces paramètres. Il existe cependant des organismes dits extrêmophiles dont les conditions de vie sont létales pour les autres organismes. Exemples :
L'étude de la variation de ces paramètres physico-chimiques sur l'activité enzymatique permet d'obtenir des informations sur le complexe ES et sur l'état de transition ES‡, et aussi sur les étapes critiques (souvent limitantes) de la réaction. |
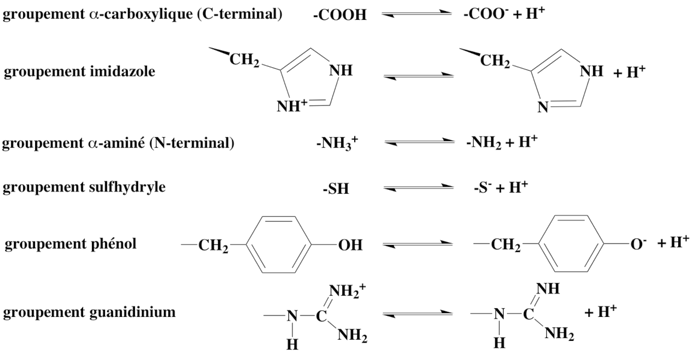
1. Effets du pH a. Incidence du pH sur la structure et la fonction des enzymes Les enzymes sont des macromolécules amphotères qui contiennent un grand nombre de groupements ionisables (acides ou basiques) portés par les 2 acides aminés situés aux extrémités N- et C-terminales et par les chaînes latérales des acides aminés qui constituent la chaîne polypeptidique. Figure ci-dessous : Equilibre d'ionisation des groupements chimiques des acides aminés des protéines.
La majorité de ces groupements étant à la surface des protéines, leur charge dépend de leur pKa et du pH de l’environnement cellulaire. |
| Groupement | pKa | ΔHionisation (kcal.mol-1) |
| α-carboxylique (C-terminal) | 3,0 - 3,2 | ± 1,5 |
| α-carboxylique (Asp ou Glu) | 3,0 - 5,0 | |
| imidazole (His) | 5,5 - 7,0 | 6,9 - 7,5 |
| α-aminé (N-terminal) | 7,5 - 8,0 | 10 - 13 |
| ε-aminé (Lys) | 9,5 - 10,6 | |
| sulfhydryle (Cys) | 8,0 - 8,5 | 6,5 - 7,0 |
| phénol (Tyr) | 9,8 - 10,5 | 6 |
| guanidinium (Arg) | 11,6 - 12,6 | 12 - 13 |
La gamme de valeurs de pKa indique que la valeur réelle est conditionnée par l'environnement physico-chimique du groupement considéré (structure tridimensionnelle et dynamique conformationnelle de l'enzyme, accessibilité au solvant). Par exemple, le groupement γ-carboxylique du Glu (acide aminé libre) a un pKa = 4 mais il se comporte comme un acide plus faible dans un environnement fortement non polaire comme l'intérieur de la structure de l'enzyme. La variation des charges des acides aminés affecte évidemment la charge nette globale des enzymes et la distribution des charges à leur surface. En conséquence, la variation du pH affecte la stabilité et la solubilité des enzymes. A des pH extrêmes, il y a un excès de charge de même signe (charges négatives portée par les groupements aminés à pH acide et charges positives portées par les groupements acides à pH alcalin). De plus, les interactions électrostatiques et les liaisons hydrogène qui ont un rôle clé (de par leur nombre notamment) dans la stabilisation de la structure tridimensionnelle des enzymes sont rompues. La dénaturation induite par le pH est un processus d'autant plus irréversible que l'enzyme est exposée à des pH extrêmes. La variation des charges des acides aminés affecte également la réactivité des groupes impliqués dans la réaction enzymatique (fixation du substrat et/ou catalyse) :
|
| Enzyme | pH optimum | Enzyme | pH optimum |
| Catalase - E.C. 1.11.1.6 | 7,0 | Myeloperoxidase - E.C. 1.11.2.2 | 4,7 - 6,0 |
| Lipase (estomac) - E.C. 3.1.1.3 | 4,0 - 5,0 | Lipase (pancréas) - E.C. 3.1.1.3 | 8,0 |
| Amylase (malt) - E.C. 3.2.1.1 | 4,6 - 5,2 | Amylase (pancréas) - E.C. 3.2.1.1 | 6,7 - 7,0 |
| Maltase - E.C. 3.2.1.20 | 6,1 - 6,8 | Invertase - E.C. 3.2.1.26 | 4,5 |
| Trypsine - E.C. 3.4.21.4 | 7,8 - 8,7 | Pepsine - E.C. 3.4.23.1 | 1,5 - 1,6 |
| Uréase - E.C. 3.5.1.5 | 7,0 | Triose-phosphate isomérase - E.C. 5.3.1.1 | 6,0 - 8,4 |
Les valeurs de pH optimal sont très variables d'une famille d’enzymes à une autre, et sont souvent liées au compartiment subcellulaire ou à l’organe (exemple : pH 8 dans le duodénum) dans lequel elles agissent. |
| Compartiment | pH | Potentiel électrique (mV) |
| Cytosol | 7,2 | 0 |
| Fluide extracellulaire | 7,4 | 30 |
| Appareil de Golgi | 6,3 | 0 |
| Lysosome | 3,5 - 5,5 | 19 |
| Mitochondrie | 8,0 | - 155 |
| Noyau | 7,2 | 0 |
| Réticulum endoplasmique | 7,2 | 0 |
| Peroxisome | 7,0 | 12 |
| Potentiel électrique relatif à celui du cytosol, à 37°C et pour une force ionique de 0,15 M. | ||
|
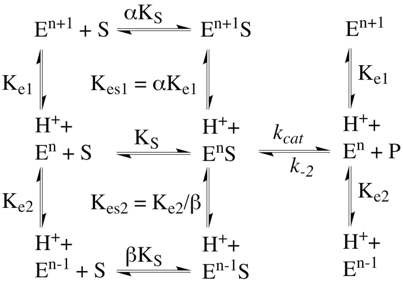
b. Equilibres de fixation du proton et formes de l'enzyme Le site actif d'une enzyme peut contenir plusieurs groupements ionisables, certains impliqués dans la fixation du substrat, d'autres dans la catalyse de la réaction enzymatique. Figure ci-dessous : Equilibres d'ionisation de groupements chimiques d'acides aminés qui contrôlent l'activité d'une enzyme. Exemple de 2 groupements impliqués dans l'activité :
En regard de la complexité tant du nombre de réactions mises en jeu que de leur formalisme, il est nécessaire de faire des simplifications justifiées :
Le facteur multiplicatif de [S]0 est la fonction de saturation en fonction du pH pour toutes les formes « ES ». Par exemple :
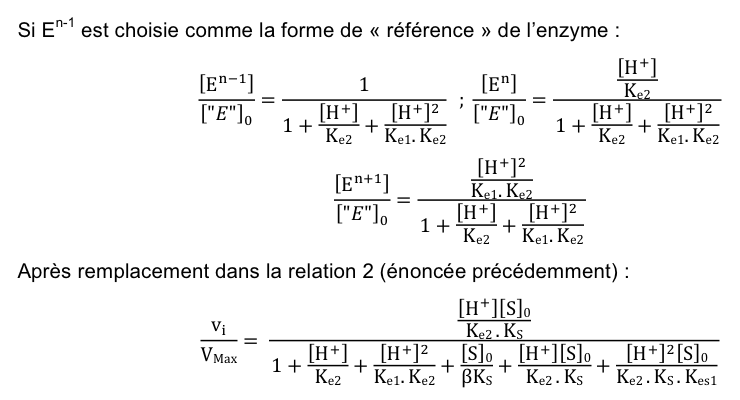
Les 6 termes du dénominateur correspondent respectivement à En-1 (forme de « référence »), En, En+1, En-1S, EnS et En+1S. Le terme ([S]0/βKS) est identique au terme ([H+][S]0Kes2/Ke2KS[H+]) car la constante d'équilibre globale entre En-1 et En-1S est la même quel que soit le chemin suivi dans le schéma réactionnel. En remplaçant par ce terme et en multipliant le numérateur et le dénominateur par (Ke2/[H+]), on aboutit à la relation 4 énoncée précédemment. En conclusion, la même équation est obtenue quelle que soit la forme de l'enzyme considérée comme « référence ». |
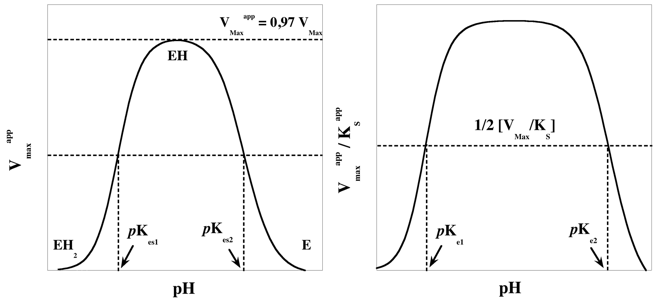
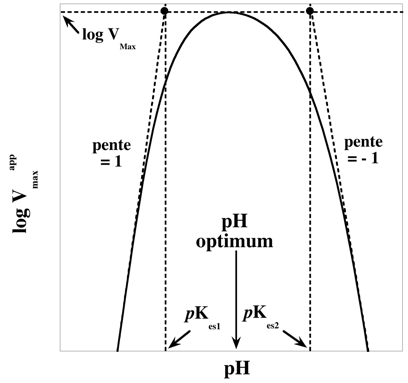
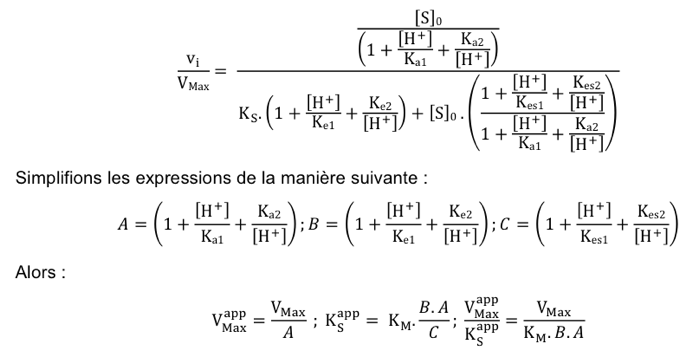
c. Formalisme des paramètres cinétiques en fonction du pH On retrouve bien évidemment la relation de Michaelis-Menten-Henri. Pour n’importe quelle valeur de pH, le formalisme est :
|
e. Cas d'un substrat qui s'ionise Si le substrat a des groupements ionisables dont les valeurs de pKa (pKa1 et pKa2) sont dans la gamme de pH étudiés, l'équation de vitesse s'écrit :
|
|
|
|
L'activité de la plupart des enzymes est optimale entre 15°C et 45°C. La vitesse des réactions chimiques non catalysées augmente de manière exponentielle avec la température. En ce qui concerne les réactions biochimiques catalysées par des enzymes, les effets de la température sont complexes. En effet, la température affecte le type et la force des interactions intramoléculaires et avec le solvant. Or ces interactions régissent la stabilité de la structure de l'enzyme donc la géométrie du site actif. Cette géométrie se répercute sur la fixation du substrat et/ou la libération du produit (effet sur les constantes d'équilibre) et/ou la réactivité des groupements impliqués dans la catalyse (effet sur la constante catalytique). a. Effet de la température sur la constante d'équilibre La constante d'équilibre Keq et la température sont liées par la relation de Jacobus Henricus van 't Hoff (1852 - 1911, Prix Nobel en 1901) :
ΔG0, ΔH0 et ΔS0 sont respectivement les variations d’énergie libre de Gibbs, d’enthalpie et d’entropie du système. T est la température absolue (K) et R est la constante des gaz parfaits (8.314 J.mol-1.K-10). En intégrant cette équation entre Keq1 et Keq2 à des températures T1 et T2 :
Cette équation (Gibbs-Helmholtz) permet de calculer la variation du pKa d'un groupement ionisable en fonction de la température (pKa = -log10app), d'où la constante 2.303). Le changement de pKeq en fonction de la température peut être conjugué à la variation en fonction du pH pour identifier les groupements impliqués dans la catalyse. On trace les graphiques VMaxapp = f(pH) et de VMaxapp / KSapp = f(pH) à différentes températures et les valeurs de pKes et de pKe sont reportées en fonction de 1/T. La pente permet de déterminer la valeur d'enthalpie ΔH0 du groupement ionisable. |
b. Energie d'activation d'une réaction et constante de vitesse
|
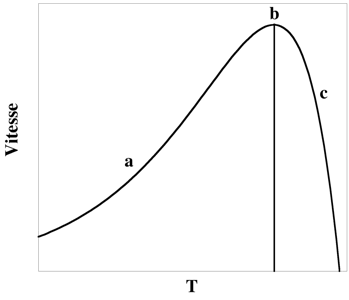
d. Cinétique de l'inactivation thermique Selon le modèle classique, l'activité enzymatique augmente jusqu'à la température optimale pour l'enzyme considérée puis diminue en raison de la dénaturation irréversible de l'enzyme. A température basse, la vitesse de dénaturation est faible. En revanche, puisque Eadénaturation > Eacatalytique, la vitesse de dénaturation augmente davantage que la vitesse de catalyse quand la température augmente. Figure ci-dessous : variation de l'activité enzymatique en fonction de la température
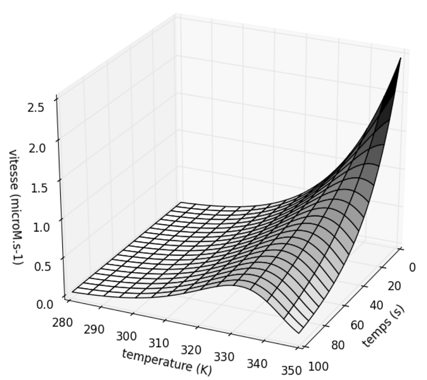
Figure ci-dessus : variation de la vitesse en fonction de la température et du temps d'incubation. Graphique tracé avec les valeurs : ΔG‡cat = 50 kJ.mol-1 ; ΔG‡inact = 96 kJ.mol-1 ; [E]0 = 10-5 M. La température optimale apparente diminue quand le temps de l'essai enzymatique augmente. Voir le script Python - Matplotlib. Le modèle précédent a été sophistiqué de diverses manières. Par exemple, en introduisant une forme intermédiaire inactive mais non dénaturée : cette forme inactive est en équilibre rapide avec la forme active et c'est la forme inactive qui subit la dénaturation irréversible.
|
| Valeurs de grandeurs thermodynamiques pour des enzymes qui suivent le modèle impliquant un intermédiaire inactif non dénaturé | ||||
| Enzyme | Classification | ΔG‡cat (kJ.mol-1) | ΔG‡inact (kJ.mol-1) | ΔHeq (kJ.mol-1) |
| Glutamate déshydrogénase | E.C. 1.4.1.2 | 63 | 93 | 264 |
| Dihydrofolate réductase | E.C. 1.5.1.3 | 67 | 93 | 104 |
| γ-Glutamyl transférase | E.C. 2.3.2.2 | 63 | 98 | 109 |
| Citrate synthase | E.C. 2.3.3.1 | 77 | 103 | 164 |
| Phosphatase alcaline | E.C. 3.1.3.1 | 72 | 99 | 305 |
| Phosphatase acide | E.C. 3.1.3.2 | 79 | 95 | 142 |
| β-Glucosidase | E.C. 3.2.1.21 | 88 | 103 | 149 |
| Fumarase | E.C. 4.2.1.2 | 60 | 92 | 378 |
| Source : Daniel & Danson(2013) | ||||
|
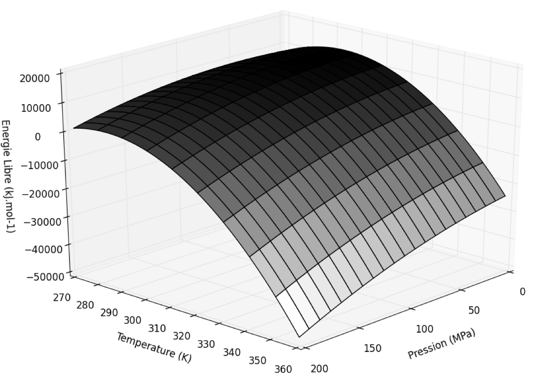
Des pressions de 1000 à 2000 bars (pression atmosphérique : P0 = 1 atm = 1.013 bar = 1.013 105 Pa) dissocient les protéines oligomèriques (structure quaternaire) en sous-unités, des pressions de 4000 à 8000 bars dénaturent les structures tertiaires et les structures secondaires ne sont affectées qu'à partir de pressions de 10000 bars. Les changements de pression affectent les réactions enzymatiques : une augmentation de la pression d'un facteur 1000 peut résulter en une diminution d'un facteur 2 de kcat et/ou de KM.
La pression peut diminuer l'activité enzymatique jusqu'à inactivation complète à 2000 bars : cette inactivation est associée à des modifications de l'étape limitante provoquées par une hydratation accrue du site actif et/ou des changements conformationnels mineurs du site actif qui ont lieu lors de la compression. De telles pressions n'ont rien à voir avec les conditions physiologiques même extrêmes. Elles ont cependant un intérêt majeur du point de vue théorique car elles permettent d'obtenir des informations concernant un état difficile à caractériser : l'état de transition. En conséquence, on obtient des informations quant aux différences entre le complexe ES (le substrat fixé de manière lâche à l'enzyme) et l'état de transition (ES‡). |
a. Effet de la pression sur la constante d'équilibre et la constante catalytique : volume d'activation Selon le principe de Henry Louis Le Chatelier (1850 - 1936), toute pression change la répartition entre les équilibres observés à la pression atmosphérique. Ces changements de distribution obéissent à la relation :
|
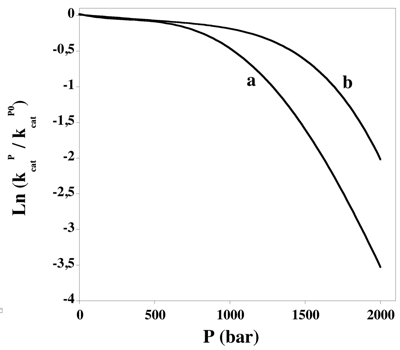
b. Ecart à la linéarité de la variation de la constante catalytique en fonction de la pression Pour de nombreuses enzymes, les données expérimentales ne correspondent pas à une droite (relation 2 - paragraphe précédent). Cette non-linéarité peut être interprétée en termes d'interdépendance entre la pression et d'autres paramètres (pH, température, propriétés physique du solvant - l'eau dans les conditions physiologiques), de dépliement de l'enzyme induit par la pression, de changements de compressibilité ou de changements de vitesse.
Figure ci-dessus : variation non linéaire de la constante catalytique en fonction de la pression. (a) : T = 37°C; (b) : T = 25°C. Les données sont lissées avec un polynôme de degré 4. Valeurs issues de Decaneto et al. (2015) avec la métalloprotéase 14 de la matrice extracellulaire (E.C. 3.4.24.80).
Exemple : la butyrylcholine estérase de l’homme (E.C. 3.1.1.8) subit une augmentation du volume hydrodynamique au-dessus d'une pression critique. Cette augmentation reflète probablement l'intercalation de molécules d'eau entre les éléments de structure secondaire, la structure de la protéine "gonflant comme une éponge gorgée d'eau". |
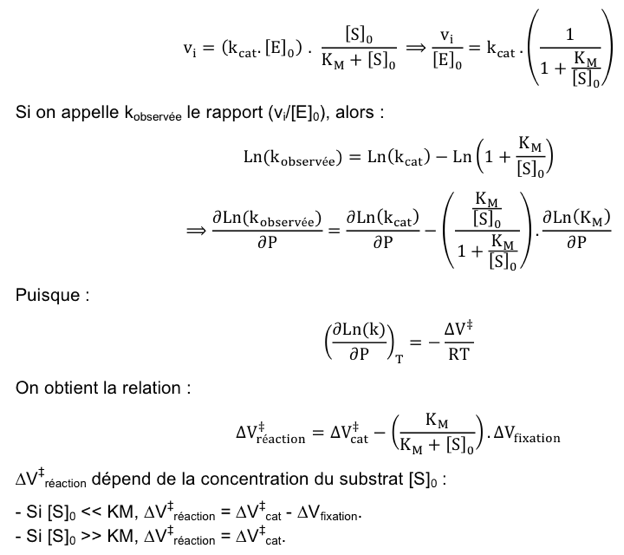
c. Effet de la pression sur la fixation du substrat sur l'enzyme La réaction enzymatique inclue une étape initiale de fixation du substrat sur l'enzyme (E+S -> ES), régie par la constante KM. Le volume d'activation de la réaction enzymatique (ΔV‡réaction) a donc 2 contributions :
La relation générale de Briggs-Haldane s'écrit :
|
|
L'expression la plus simple de la force ionique I est :
Où Ci est la concentration molaire de l'ion i et zi sa charge. Exemple : la force ionique d'une solution de CaCl2 0,1 M = 0.5 x [(0,1 x 22) + (2 x 0,1 x (-1)2)] = 0,3 M. Une enzyme n'est active que si elle est solubilisée dans le milieu où elle agit. En solution, chaque molécule d'enzyme est hydratée, c'est-à-dire en interaction avec des molécules d'eau. Si on introduit un sel dans cette solution, il est aussi hydraté. Cependant, si ce sel est à une concentration beaucoup plus grande que l'enzyme, il monopolise toutes les molécules d'eau et l'enzyme n'a plus d'enveloppe d'hydratation : elle est séparée du solvant aqueux. C'est le processus de relargage ou "salting-out". En dehors de l'étude des propriétés catalytiques des enzymes, cette modification de la solubilité des enzymes (des protéines en général) est aussi utilisée pour purifier les enzymes ou les protéger de la dénaturation (par exemple). Les interactions [enzyme - eau] et [sel - eau] (voire [enzyme - sel]) dépendent :
La relation entre la solubilité d'une enzyme et la force ionique s'écrit :
Où S0 est la solubilité de l'enzyme en absence de sel, S la solubilité de l'enzyme en présence du sel étudié, A est une constante (enzyme, pH, température), za et zb sont les charges des ions du sel et KS est le coefficient de relargage du sel.
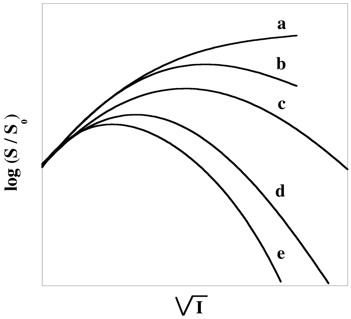
Figure ci-dessus : effet du type de sel (racine carrée de la force ionique) sur la solubilité de l'hémoglobine. (a) : NaCl; (b) : KCl; (c) : MgSO4; (d) : (NH4)2SO4; (e) : Na2SO4. La solubilité (mesurée à 25°C) est en g.(1000 g H20)-1. Valeurs adaptées de Green (1932). |
b. Effet sur l'activité des enzymes Outre le pH, la charge de la chaîne latérale des acides aminés dépend de la force ionique : c'est donc un paramètre qui affecte l'activité enzymatique. C'est particulièrement probant quand la catalyse dépend du mouvement les unes vers les autres de molécules chargées : la fixation des substrats chargés dans le site catalytique et leur mouvement sont influencés par la composition en ions du milieu. La constante de vitesse d'une réaction dans des conditions standard (k0) est liée à la constante de vitesse de cette réaction dans d'autres conditions (k) par la relation :
Où kcatI est la constante catalytique de la réaction à la force ionique étudiée, kcatI0 est la constante catalytique à une force ionique = 0, za et zb sont les charges des molécules réactives (exemples : enzyme et substrat, deux groupements réactifs de l'enzyme, …). Cette relation indique que :
Exemple : l'étape qui contrôle la vitesse de catalyse par la chymotrypsine implique le rapprochement de 2 groupes chargés positivement, His57 et Arg145. L'augmentation de la force ionique du milieu réactionnel augmente de manière significative la valeur de kcat. |
| 10. Liens Internet et références bibliographiques |
Arrhenius S.A. (1889) "Über die Dissociationswärme und den Einfluß der Temperatur auf den Dissociationsgrad der Elektrolyte" Z. Phys. Chem. 4, 96 - 116 Arrhenius S.A. (1889) "Über die Reaktionsgeschwindigkeit bei der Inversion von Rohrzucker durch Säuren" Z. Phys. Chem. 4, 226 - 248 Green A.A. (1932) "The solubility of hemoglobin in solutions of chlorides and sulfates of varying concentrations" J. Biol. Chem. 95, 47 - 66 Eyring H. (1935) "The activated complex in chemical reactions" J. Chem. Phys.3, 107 - 115 Kramers H.A. (1940) "Brownian motion in a field of force and the diffusion model of chemical reactions" Physica 7, 284 - 304 Glasstone et al. (1941) "The theory of rate processes", McGraw-Hill Book Co., Inc., New York City |
Dixon M. & Webb E.C. (1964) "Enzymes" 2nd Ed., Academic Press, Chap. 4, 116 - 166 Hawley S.A (1971) "Reversible pressure-temperature denaturation of chymotrypsinogen" Biochemistry 10, 2436 - 2442 Segel I.H. (1975) "Enzyme kinetics", John Wiley, New York |
Morild E. (1977) "Pressure variation of enzymatic reaction rates : yeast and liver alcohol dehydrogenase" Biophys. Chem. 6, 351 - 362 Antonini E. & Ascenzi P. (1981) "The mechanism of trypsin catalysis at low pH. Proposal for a structural model" J. Biol. Chem. 256, 12449 - 12455 Norby J.G. & Esmann M. (1997) "The effect of ionic strength and specific anions on substrate binding and hydrolytic activities of Na,K-ATPase" J. Gen. Physiol. 109, 555 - 570 Cornish-Bowden et al. (2005) « Cinétique enzymatique », Collection Grenoble Sciences, EDP Sciences Masson P. & Balny C. (2005) "Linear and non-linear pressure dependence of enzyme catalytic parameters" Biochim. Biophys. Acta 1724, 440 - 450 |
Wiedersich et al. (2008) "Temperature and pressure dependence of protein stability: the engineered fluorescein-binding lipocalin FluA shows an elliptic phase diagram" Proc. Natl. Acad. Sci. 105, 5756 - 5761 Struvay C. & Feller G. (2012) "Optimization to low temperature activity in psychrophilic enzymes" Int. J. Mol. Sci. 13, 11643 - 11665 Huang X. & Hernick M. (2012) "Examination of mechanism of N-Acetyl-1-d-myo-inosityl-2-amino-2-deoxy-α-d-glucopyranoside deacetylase (MshB) reveals unexpected role for dynamic tyrosine" J. Biol. Chem. 287, 10424 - 10434 Daniel R.M. & Danson M.J. (2013) "Temperature and the catalytic activity of enzymes : a fresh understanding" FEBS Letters 587, 2738 – 2743 |
Andrea Salis A. & Ninhamb B.W. (2014) "Models and mechanisms of Hofmeister effects in electrolyte solutions, and colloid and protein systems revisited" Chem. Soc. Rev. 7358 - 7377 Decaneto et al. (2015) "Pressure and temperature effects on the activity and structure of the catalytic domain of human MT1-MMP" Biophys. J. 109, 2371 - 2381 DeLong et al. (2017) "The combined effects of reactant kinetics and enzyme stability explain the temperature dependence of metabolic rates" Ecol. Evol. 7, 3940 - 3950 van der Kamp et al. (2018) "Dynamical origins of heat capacity changes in enzyme-catalysed reactions" Nat. Commun. 9, 1177 |
![]()