| Etude des protéines LEA ("Late Embryogenesis Abundant Proteins" - LEAP) |
| Tweet |
|
|
1. Mise en évidence d'un motif 2. Affinement du motif 3. Illustration du principe de la recherche de similarité locale avec BLAST 4. Recherche de protéines possédant le(s) motif(s)
|
5. PHI-BLAST 6. Recherche de motifs répétés 7. Caractéristiques structurales des LEA 8. Recherche du maximum de séquences de LEA 9. Liens Internet et références bibliographiques |
|
"ExPASy Proteomics tools" : Ensemble d'applications pour l'analyse de séquences peptidiques. "Sequence Manipulation Suite" : Ensemble d'applications Java pour l'analyse de séquences d'ADN et de protéines. |
|
1. Mise en évidence d'un motif a. Chercher un programme de traduction de séquences nucléotidiques. b. Obtenez la traduction sur les 6 phases de la séquence nucléotidique de la protéine 1 / (ou Fichier format .rtf) c. Motifs à repérer : SSSEDD et/ou KIKEKL. Récupérer le fichier FASTA de la protéine qui vous semble le plus logiquement correspondre à la séquence de la protéine 1. |
|
a. Effectuer le même travail avec les séquences nucléotidiques suivantes. [Remarque : les séquences protéiques issues de la traduction sont inclues dans ce fichier]. b. Avec un programme d'alignement multiple, aligner les séquences protéiques traduites afin de mettre en évidence un ou des motifs communs à ces séquences de protéines. Tester l'un des programmes : Clustal Omega / Muscle / T-coffee / MAFFT.
c. Récupérez les séquences FASTA de : CAJ56060, AAD02258, CAA33364, CAJ56055, CAA68765, AAB05927, AAN08718, BAD13498, BAD86644. Alignez les avec le séquences traduites et le programme MULTALIN. Que peut-on conclure ? |
2b. Affinement du motif - suite a. Aller à "InterPro" et soumettre la séquence FASTA du fichier N° accession AAD02258. Résultat : en principe pluseurs graphiques sont obtenus. Cliquer sur chacun des liens "PS00823" et "PS00315" à droite du graphique du milieu "IPR030513 / Dehydrin, conserved site". Enregistrer l'expression régulière du motif signature ("pattern motif"). Pattern motif N° PS00315 Pattern motif N° PS00823 |
| Voir le fichier "aide" |
|
3. Illustration du principe de la recherche de similarité locale avec BLAST Effectuer une recherche de similarité locale avec BLAST à partir de :
Les résultats sont-ils si étonnants vu le principe de BLAST ? |
4. Recherche de protéines possédant le(s) motif(s) : PSI-BLAST En incluant dans la recherche les séquences des protéines "proches" de la séquence requête, la recherche avec PSI-BLAST ("Position Specific Iterated BLAST") est beaucoup plus sensible que celle avec BLAST pour trouver des "parents lointains" de la séquence requête.
Le programme PSI-BLAST est aussi utilisé pour les séquences nucléotidiques. |
|
Faire une recherche avec PSI-BLAST ("Position-Specific Iterated BLAST") avec la séquence requête P22239. Quelle est cette protéine ? Effectuer 2 itérations et commenter l'évolution des résultats. Remarque : le paramètre "Inclusion threshold" = 0.005 par défaut. |
| Voir le fichier "aide" |
Récupérer les séquences les plus similaires du résultat de PSI-BLAST. Aller à MULTALIN. Effectuer plusieurs alignements en modifiant le choix de la matrice et les valeurs des gaps.
Aller à LEAPdb ("Late Embryogenesis Abundant Proteins Database" - base de données dédiée aux protéines de la famille "LEA") et effectuer un BLAST avec la séquence ADT65201.
|
|
C'est un programme adapté à :
PSI-BLAST est le programme dérivé de BLAST le plus sensible ce qui en fait un excellent outil pour trouver des protéines trés distantes.
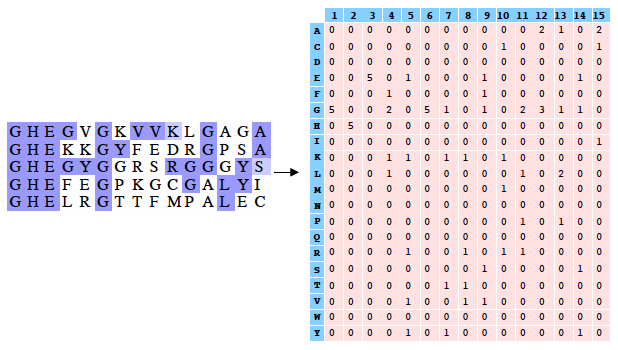
Exemple (très simple) d'alignement multiple de 2 séquences de 4 acides aminés : DWKD Le profil correspondant (en probabilités) : 1 2 3 4 D 1.0 0.0 0.0 0.5 G 0.0 0.0 0.0 0.5 K 0.0 0.0 0.5 0.0 N 0.0 0.0 0.5 0.0 W 0.0 1.0 0.0 0.0 Ce qui ce signifie :
L'utilisation d'un profil permet une recherche beaucoup plus sensible de séquences homologues éloignées que l'utilisation d'une séquence seule car le profil correspond à une information sur la variabilité des différentes positions parmi les protéines connues. En contrepartie un profil est moins spécifique qu'une simple séquence seule. Si on utilise PSI-BLAST sur un sous ensemble particulier de séquences, il est probable que l'on ne trouve pas tous les homologues, surtout si leur séquence est peu conservée par rapport à la séquence requête. Pour améliorer la sensibilité de détection des homologues, il est préférable d'effectuer un alignement avec PSI-BLAST sur une banque de séquences plus grande. Mais la sensibilité est diminuée si la banque de données est trop grande puisque la fréquence d'observation d'un score particulier (la "E-value") augmente avec la taille de la banque de données. Or, pour un alignement de 2 séquences, plus le score est petit, plus la probabilité que ces 2 séquences soient homologues est grande. Il est donc préférable de chercher d'abord dans une banque "nettoyée" ("curated") comme la base de données non-redondante "nr" où toutes les séquences identiques ont été éliminées sauf un exemplaire. Si plusieurs séquences sont dans cette banque, on peut calculer un profil et l'utiliser pour effectuer une nouvelle recherche dans ce sous ensemble. On augmente ainsi la sensibilité de la recherche d'homologues.
Les programmes des familles Fasta et BLAST sont des heuristiques qui réduisent le facteur temps en "sacrifiant" un peu de sensibilité. L'un et l'autre simplifient le problème :
|
|
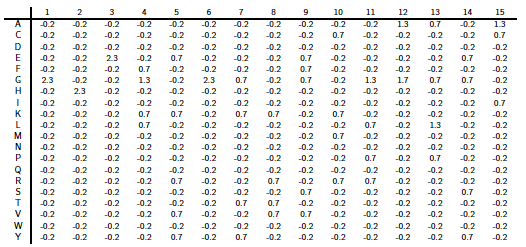
Ci-dessous : la matrice PSSM "Position Specific Scoring Matrice" complète calculée à partir de l'exemple précédent.
|
|
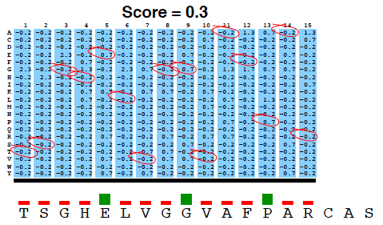
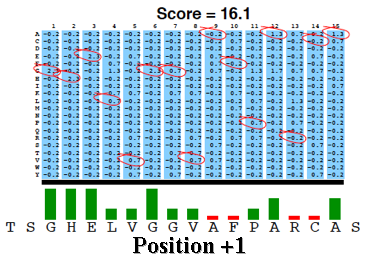
La matrice PSSM est ensuite appliquée à la séquence requête en utilisant une "fenêtre glissante".
A chaque position, un score PSSM est calculé en sommant les scores de toutes les colonnes. Le plus haut score est retenu.
Source des figures : Pagni M. (2003) "An introduction to Patterns, Profiles, HMMs and PSI-BLAST" / SIB Course |
| Conclusion PSSM : à utiliser pour modéliser de courtes régions avec une forte variabilité mais de longueurs constantes. | |
| Avantages | Inconvénients |
|
|
| Exemples d'outils | Exemples de bases de données |
|
5. PHI-BLAST ("Pattern Hit Initiated BLAST") Ce programme prend en entrée une séquence requête protéique et un motif défini par une expression régulière. PHI-BLAST est adapté à la recherche de séquences protéiques qui contiennent un motif spécifié par l'utilisateur (fenêtre "PHI pattern" de la section "Algorithm") ET sont similaires à la séquence requête (fenêtre "Search") dans le voisinage proche du motif. La syntaxe du motif doit suivre la syntaxe de PROSITE. Exemple : <A-x-[ST](2)-x(0,1)-{V}
Exemple 1 de syntaxe de motif : [KR]-[LIM]-K-[DE]-K-[LIM]-P-G Exemple 2 de syntaxe de motif : S(4)-[SD]-[DE]-x-[DE]-[GVE]-x(1,7)-[GE]-x(0,2)-[KR](4) Faire une recherche avec PHI-BLAST sur la base de un (ou deux) motifs communs à la famille de protéines étudiée. Attention : vérifier la syntaxe des motifs que vous choisissez. |
6. Recherche de motifs répétés a. 1ère partie Récupérer la séquence FASTA de "AAC05921" dans LEAPdb. Aller à la base de données PROSITE ("Database of protein domains, families and functional sites"). Remarque : l'EBI propose également un programme ("InterProScan sequence search").
|
| Voir les motifs des 12 classes de LEAP |
Aller à la page du motif "PS00823". Une déhydrine d'interêt pour l'équipe "Mitostress" de l'IRHS - Angers ne possède pas l'un des motifs. De quel végétal s'agit-il ? Retrouver les séquences de déhydrines de
ce végétal et aligner avec d'autres déhydrines spécifiques. |
|
b. 2ème partie
|
|
7. Caractéristiques structurales des LEA Certaines LEA ont une caractéristique structurale. Récupérer la séquence FASTA de "AAC05921" dans LEAPDB. Aller à l'ancienne version de : "DisProt: Database of Disordered Proteins". Quel est le but de cette base de données ? Remarque : voir la version récente de DisProt. Choisir l'item "Disorder Predictors" (menu de gauche). Tester plusieurs programmes de prédiction de sructure, en particulier "Fold Index".
|
|
8. Recherche de séquences de LEA Aller au NCBI et taper "late embryogenesis abundant OR Lea OR dehydrin" en choisissant "Protein". Examiner le résultat. Elaborer un crible de plus en plus précis et exhaustif afin de réduire le nombre de résultats aux seules LEA. Pour celà s'inspirer du fichier : "1RequeteRemplirLEA". |
| 9. Liens Internet et références bibliographiques |
|
LEAPDB("Late Embryogenesis Abundant Proteins Database") |
|
|
Logiel d'alignement "Multalin" |
|
| Seqret (EMBOSS) - biosequence conversion tool | |
|
Grelet et al. (2005) |
|