| La bioinformatique |
| Tweet |
|
|
1. La bioinformatique : définition, description, démarche et principales étapes 2. Deux types de molécules support de la bioinformation : les acides nucléiques et les protéines 3. Deux types de bioinformation : la séquence des nucléotides et la séquence des acides aminés 4. Le séquençage du génome humain |
5. Le stockage de la bioinformation : les banques de données 6. La structuration de la bioinformation : fichiers et formats 7. Exemples d'algorithmes et de programmes en bioinformatique 8. Exemple d'analyse bioinformatique d'une enzyme : la glutamate déshydrogénase (GDH) 9. Propositions d'emplois en bioinformatique |
|
1. La bioinformatique : définition, description, démarche et principales étapes Définition : La bioinformatique est l'analyse de la bioinformation. La bioinformation est l'information liée aux molécules biologiques : leurs structures, leurs fonctions, leurs liens de "parenté", leurs interactions et leur intégration dans la cellule. Divers domaines d'études permettent d'obtenir cette bioinformation : la génomique structurale, la génomique fonctionnelle, la protéomique, la détermination de la structure spatiale des molécules biologiques, la modélisation moléculaire ... Description : C'est une discipline récente (quelques dizaines d'années). C'est une discipline "hybride" (au même titre que la biochimie ou la biophysique) : elle est fondée sur des concepts et des formalismes issus de la biologie, de l'informatique, des mathématiques et de la physique. C'est une discipline qui utilise toutes les potentialités de traitement de l'informatique : modèles théoriques, algorithmes et programmes, ordinateurs, réseau Internet, bases de données, langages, ... Démarche 1. Compilation et organisation des données biologiques dans des banques de données : ces banques sont soit généralistes (elles contiennent le plus d'information possible sans expertise particulière de l'information déposée), soit spécialisées dans un domaine autour de thèmes précis. 2. Traitements systématiques des données : l'objectif principal est de repérer et de caractériser une fonction et/ou une structure biologique importante. Les résultats de ces traitements constituent de nouvelles données biologiques obtenues "in silico". 3. Elaboration de stratégies :
Voir les grandes étapes de l'évolution de la bioinformatique. |
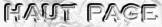
| 2. Deux types de molécules support de la bioinformation : les acides nucléiques et les protéines |
|
ADN : Acide DésoxyriboNucléique
On distingue :
|
|
ARN : Acide RiboNucléique
On distingue :
|
Protéine
|
|
3. Deux types de bioinformation : la séquence des nucléotides et la séquence des acides aminés Les séquences constituent l'un des principaux types de bioinformation qu'analyse la bioinformatique. Les chaînes nucléotidiques (ADN, ARN) et les chaînes polypeptidiques (protéines) sont des polymères d'unités élémentaires :
Elles possèdent 2 extrémités distinctes et sont donc orientées :
En conséquence :
L'obtention des séquences
|
| Exemples d'autres types de bioinformation (directe ou obtenue "in silico") | |
| Les structures tridimensionnelles des protéines et aussi, malgré leur nombre plus restreint, des acides nucléiques (en particulier les ARN de transfert). | Protein Data Bank |
| Les données obtenues en protéomique (gels d'électrophorèse bidimensionnel). | SWISS-2DPAGE |
| Le changement d'un nucléotide dans un gène quelconque ("Single Nucleotide Polymorphism"). | SNP |
| La taxonomie (classification) des organismes. | Taxonomy |
| L'ontologie : l'organisation hiérarchique de la connaissance sur un ensemble d'objets par leur regroupement en sous-catégories suivant leurs caractéristiques essentielles. | GO |
| Les données bibliographiques (diffusion des résultats de la recherche par les articles). | PubMed |
|
4. Le séquençage du génome humain La molécule d'ADN est le support biologique de l'information héréditaire. Cette information est transmise par la copie conforme de cette molécule. C'est une macromolécule formée par l'enchaînement de milliers, de millions ou de milliards (dans le cas de l'homme) de nucléotides. L'extension complète de l'ADN humain formerait un fil de plus d'1,2 m. Super-enroulement de l'ADN dans les chromosomes qui explique son extrème compacité : voir un cours sur l'épigénétique. Figure ci-dessous : une seule molécule d'ADN (long brin jaune) d'une bactérie Escherichia coli éclatée par un choc osmotique. Un fragment de la molécule d'ADN d'environ la moitié de la longueur de la bactérie correspond à environ 5000 paires de bases.
Source : L'information biologique |
| Figure ci-dessous : aperçu du déroulement du séquençage du génome humain.
Source : Nature 409, 860 - 921 |
| 1984 à 1990 |
Les pré-projets.
|
|
| 1990 | Le NIH ("National Institute of Health" - USA) et le DOE ("Department of Energy Office of science"- USA) présentent au Congrès américain le projet "Génome Humain" ("Human Genome Project" - HGP). HGP est un consortium regroupant des laboratoires de différents pays (Etats-Unis, Royaume-Uni, Japon, Allemagne, Chine et France - Génoscope). Il est financé par des fonds publiques et caritatifs. Pour éviter les problèmes liés au dépôt de brevet, les résultats du séquençage sont accessibles à tous sur internet dans les 24 heures. |
Les objectifs du HGP étaient
de :
|
| 1992 | Première phase du projet : première carte physique génétique complète (basse résolution). | Voir une belle animation décrivant le principe du séquençage (Jussieu - Génoscope). |
| 1993 | Le Généthon fournit des mega-YACs au HGP. | Le YAC ("Yeast Artificial Chromosome") est un vecteur utilisé pour cloner des fragments d'ADN jusqu'à une taille de 400 kb. Le BAC ("Bacterial Artificial Chromosome") est aussi un vecteur utilisé pour cloner des fragments d'ADN jusqu'à une taille de 300 kb. |
| 1997 | Séquençage complet du génome de la bactérie Escherichia Coli. Publication de cartes physiques génétiques à haute résolution des chromosomes humains 7 et X. | Le décryptage du génome pose la question de la brevetabilité du vivant, l'UNESCO le 11 novembre 1997 à déclaré que le génome humain est un patrimoine de l'humanité, or un patrimoine de l'humanité ne peut pas être la propriété d'un individu. Donc, une séquence d'ADN ne peut pas être brevetée. |
| 1998 | Création de la société "Celera Genomics" (USA) par Craig Venter dans le but de séquencer le génome humain en compétition avec l'HGP. Cette société a fait le choix de séquencer l'ADN de cinq personnes d'origine : africaine, asiatique, caucasienne et latino-américaine. | Une vraie course au séquençage est lancée : l'enjeu est la propriété publique ou privée du génome humain dans le but d'une exploitation commerciale des tests et des médicaments (brevets). |
| 1999 | Première séquence complète du chromosome 22 établie par HGP. | |
| 2000 |
HGP annonce 90 % du séquençage
du génome humain.
"Celera
Genomics" propose les premiers résultats du séquençage
total du génome d'une personne. Publication du génome du chromosome 21. Publication du génome complet de la mouche Drosophila melanogaster. |
Communiqué commun de Tony Blair et Bill Clinton (14 mai 2000) qui annoncent leur souhait que les résultats du séquençage soient en accès libre et que les brevets soient limités à leur exploitation industrielle et commerciale. L'entreprise "Celera Genomics" va donc devoir rendre ses résultats publics de manière trimestrielle. |
| Février 2001 |
La même
semaine, publication du brouillon initial des travaux de séquençage
du génome humain complet
par :
|
1. L'ADN humain est extrêmement hétérogène. Les gènes ne sont pas répartis uniformément sur le génome. Il existe des zones qui n'en contiennent aucun, mais possèdent des séquences répétitives. On ne connait pas encore les fonctions de ces parties de l'ADN. 2. Les résultats de la société "Celera Genomics" montrent qu'il y a plus de différences entre l'ADN des deux Caucasiens qu'entre celui d'un Africain et d'un Caucasien. Tous les êtres humains sont différents et cette différence résulte des variations entre l'ADN des individus. Ces variations correspondent au changement d'un nucléotide dans un gène quelconque que l'on appelle un "Single Nucleotide Polymorphism" (SNP). Les SNPs représentent 0,1% de différence entre deux génomes (plus de 1,4 millions de SNP ont été identifiés). Ils sont particulièrement intéressants pour la médecine et l'industrie pharmaceutique (détermination de l'origine de nombreuses maladies, développement de tests de prédisposition aux maladies, synthèse de médicaments en fonction de la sensibilité génétique). 3. Le gène ZNF217 est identifié dans le chromosome 20. Ce gène apparaît en nombre croissant de copies dans beaucoup de tumeurs. Il jouerait un rôle dans le cancer du sein. |
|
5. Le stockage de la bioinformation : les banques de données Les fichiers contenant l'information biologique sous la forme de séquences est l'élément central autour duquel les banques de données se sont constituées. Il existe un grand nombre de bases de données d'intérêt biologique. On peut distinguer :
Exemple de grandes banques généralistes :
Ces trois banques s'échangent systématiquement leur contenu depuis 1987 et adoptent un système de conventions communes (The DDBJ/EMBL/GenBank Feature Table Definition).
Exemple de banques spécialisées :
|
|
8. Exemple d'analyse bioinformatique d'une enzyme : la glutamate déshydrogénase (GDH) On peut considérer la première réaction d'assimilation de l'azote (sous forme d'ammoniac) par la glutamate déshydrogénase (GDH) comme un point d'entrée dans le métabolisme azoté. L'atome d'azote est à l'origine de la fonction α-aminée des acides aminés selon la réaction : NH3+ + α-cétoglutarate + NAD(P)H + H+ <=> glutamate + NAD(P)+ Il existe trois isoformes de GDH :
La GDH4 joue peut-être un rôle clé dans l'assimilation de l'azote. Or ce rôle n'a pas encore été démontré, notamment chez les plantes. Par ailleurs, on ne dispose d'aucune information concernant la structure de la GDH4. La bioinformatique permet l'étude prospective de la relation structure - fonction de la GDH. |
![]()