Réseau d'interactions protéine-protéine - Cellules individualisées ("Single-Cell"). |
| Voir l'ensemble de ressources dédiés aux interactions entre macromolécules biologiques. |
Exercice 1 Soit une cellule d'un volume de 103 µm3 avec une concentration totale de protéines = 5 µM :
|
a. Concentration totale de protéines : 5 µM (5 10-6 moles.L-1) dans 1 cellule de volume 1 pL |
c. Nombre de molécules d'une protéine d'un type donné : x = (5 10-18 moles . 6,022 1023 molécules / 1 mole) Et : (3 106 molécules / 104 types de protéines) ≈ 300 molécules d'une protéine d'un type donné. |
b. Nombre d'Avogadro (N) : 1 mole -> 6,022 1023 molécules |
Exercice 2 Aller au site "STRING exercises" : faire l'exercice 1. Coller "INSR" dans la fenêtre "Protein Name" et choisir "Homo sapiens". Puis "SEARCH" puis "CONTINUE". Analyser le réseau créé en explorant les options des différentes fenêtres en bas. En particulier :
|
Exercice 3 : représentation de réseau avec Cytoscape a. Aller aux réseaux analysables en ligne de NDEX. Choisir l'exemple "PANCREATIC BETA CELL" [passer "Skip" & "Got it"]. Qu'est-ce que STAT3 ?
|
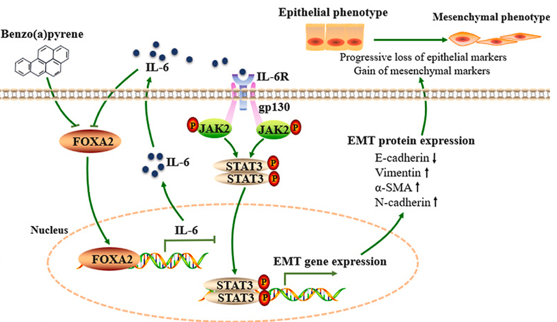
b. Trouver un article scientifique décrivant un lien entre STAT3 et FOXA2. Voir l'article Tang et al. (2024) "Mediation of FOXA2/IL-6/IL-6R/STAT3 signaling pathway mediates benzo[a]pyrene-induced airway epithelial mesenchymal transformation in asthma" Environ. Pollut. 357, 124384. Le benzo[a]pyrène (BaP) est un polluant toxique qui augmente l'incidence et la gravité de l'asthme. Après action du BaP :
Le BaP active la voie de signalisation [IL-6 / IL-6R (récepteur de l'interleukine 6 / STAT3] pour favoriser la TME des voies respiratoires dans l'asthme.
Source : Tang et al. (2024) Autre article : Hao et al. (2014) "Mycoplasma pneumoniae modulates STAT3-STAT6/EGFR-FOXA2 signaling to induce overexpression of airway mucins" Infect. Immun. 82, 5246 - 5255 |
c. Lancer le programme CYTOSCAPE. Revenir à la page de NDEX. Cliquer sur l'icône orange On peut afficher le réseau dans une fenêtre séparée en cliquant sur l'icône "Detach View" (flèche blanche vers le haut dans un petit cadre noir, en bas de la fenêtre qui affiche le réseau).
|
d. Cliquer sur le noeud "STAT3" puis sur l'icône représentant "2 maisons" ("First Neighbors of Selected Nodes") en haut. Déterminer les plus proches voisins de STAT3 : SHH, SOX2, FGF8, FOXA2 et MSX1. |
e. Regroupement des nœuds
Recommencer avec les 3 autres sous-réseaux (nœuds en colonnes) du réseau. f. Modification de l'apparence du réseau
Essayer de reproduire un réseau ayant une apparence comme celle présentée en exemple ci-dessous (1er = "Default" / 2è = "Gradient1" ) ou toute autre apparence selon l'inspiration.
Sauvegarder la figure au format et à la taille désirés avec le menu |
Exercice 4 Voir le paragraphe du cours : "4. Démarche pour la construction de réseaux d'interactions". Aller à l'exercice en ligne de l'EBI sur l'interactomique. Remplir la matrice d'adjacence ("the adjacency matrix") du graphe proposé. |
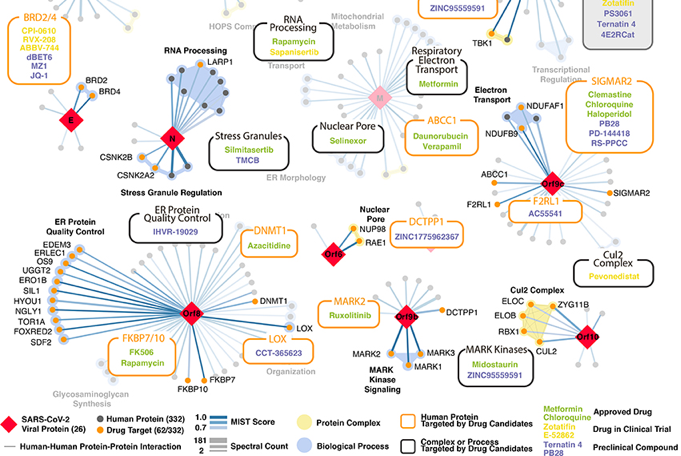
Exercice 5 Analyser le paragraphe "Identification of existing drugs targeting SARS-CoV-2 human host factors" et la figure 5 de l'article Gordon et al. (2020) "A SARS-CoV-2 Protein Interaction Map Reveals Targets for Drug-Repurposing" Nature 583, 459 – 468. Des ligands interagissant avec certaines protéines humaines ont été recherchés afin de perturber l'interactome entre ces protéines et celles du virus SARS-CoV-2. Les molécules ont été classées par ordre de priorité en fonction :
Source : Figure 5 "Drug-human target network" - Gordon et al. (2020)
Sur les 332 cibles humaines qui interagissent avec les protéines d'appât viral avec une signification élevée, 63 cibles possèdent 69 [médicaments/IND/molécules précliniques] qui les modulent et sont intégrées au réseau d'interactions protéiques. Parmi ces molécules, des ligands des récepteurs sigma1 et sigma2 ont été testés : halopéridol, PB28, PD-144418 et hydroxychloroquine (essais cliniques chez des patients atteint par la COVID-19). La zotatifine (IC90 = 37 nM) et la molécule PB28 (IC90 = 278 nM) inhibent puissamment le virus SARS-CoV-2 :
|
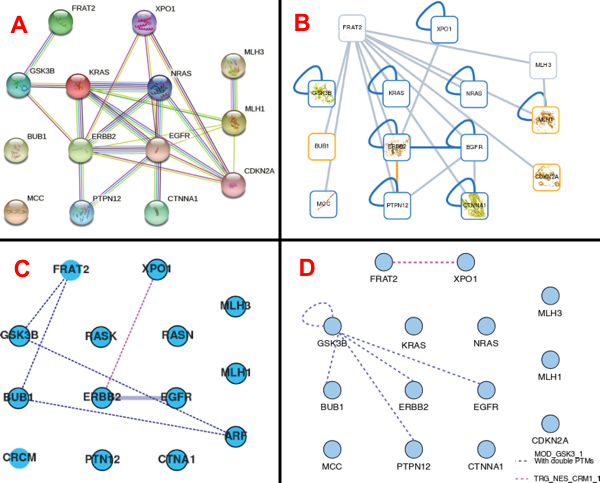
Exercice 6 a. Analyser la figure 3 de l'article Reys & Labesse (2022) "SLiMAn: An Integrative Web Server for Exploring Short Linear Motif-Mediated Interactions in Interactomes" J. Proteome Res. 21, 1654 - 1663. De très nombreuses interactions protéine-protéine impliquent des domaines qui contiennent des motifs appelés "motifs linéaires courts" ("Short Linear Motifs" - SLiM) :
La figure ci-dessous représente 4 réseaux incluant 13 protéines (mentionnées dans la base de données BioGRID) qui interagissent avec la protéine FRAT2. FRAT2 inhibe la protéine-kinase GSK-3 ("Glycogen Synthase Kinase-3") et régule positivement la voie de signalisation Wnt en stabilisant la β-caténine grâce à l'association avec GSK-3.
Source : Reys & Labesse (2022) Réseau A : généré avec les données d'interactions de la base de données d'interactions STRING. Réseau B : Idem à partir de la base de données Interactome3D. Réseau C : Idem à partir de la base de données Proteo3DNet.
Réseau D : Idem à partir de la base de données SLiMAn.
|
b. Aller à BioGRID.
Réponses : 26 protéines. Les données de l'article sont antérieures à 06/2022 (date de publication de l'article). |
Exercice 7 - "Single-Cell" Analyser et décrire la figure 1 de l'article Clark et al. (2023) "Microfluidics-free single-cell genomics with templated emulsification" Nat. Biotechnol. 41, 1557 - 1566 La technique de séquençage instantané par répartition des particules ("Particle-templated instant partition sequencing" - PIP-seq) permet l'encapsulation de cellules dans des gouttelettes en utilisant la taille de billes pour contrôler le volume des gouttelettes :
Par exemple, avec un taux de collision de 6% incluant les doublets de cellules et la réutilisation des codes-barres, l'émulsification à base de particules dans des tubes de différents volumes génère des émulsions monodispersées capables d'encapsuler les séquences contenant les codes-barres. Illustration dans la figure du haut ci-dessous :
Figure du bas : la technique PIP-seq est également adaptable aux formats de plaques à 96, 384 et 1536 puits.
Source : Clark et al. (2023)
|
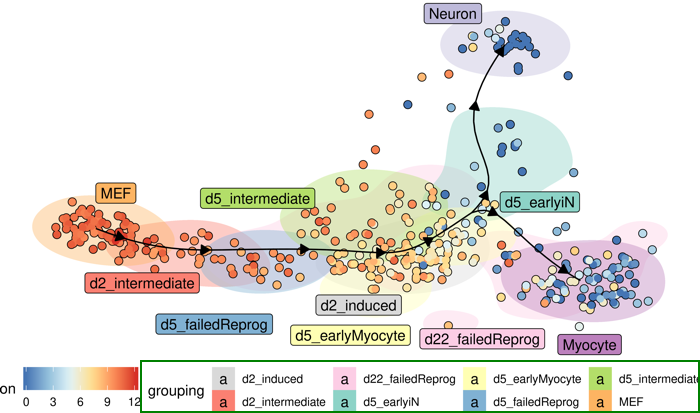
Exercice 8 - "Single-Cell trajectories" a. Rappeler la notion de trajectoires. Expliquer la signification des bifurcations et des branches. La grande diversité cellulaire résulte du caractère asynchrone de l'évolution des cellules et de l'ensemble des processus biologiques. L'analyse bioinformatique de l'inférence des trajectoires permet de décrire la progression de chaque cellule individualisée de chaque type de cellules au cours des processus biologiques qui impliquent une évolution de ces cellules (différenciation, développement, processus pathologique, ...). Les trajectoires mettent en évidence des points de ramifications où le devenir des cellules diverge : ces ramifications traduisent donc des décisions cruciales, à certains stades de ces processus, qui déterminent des destins cellulaires distincts. |
b. La figure suivante est la visualisation des trajectoires après réduction de dimensionnalité et regroupement de 3 types de cellules : fibroblaste embryonnaire de souris (MEF), neurone et myocyte. Ces données résultent de l'analyse RNAseq de cellules individualisées à plusieurs stades cellulaires, afin de décrypter les processus de reprogrammation des MEF quand ils sont différenciés en neurones et myocytes.
Source de la figure : Dynverse & Cannoodt R. (2019) En effet, en utilisant 3 facteurs de transcription (Ascl1, Pou3f2/Brn2 et Myt1l) qui jouent un rôle clé dans la différentiation des neurones, les MEF sont reprogrammés en cellules neuronales induites ("induced neuronal (iN) cells").
|
| Exercice 9 | |
| Assertions | Réponses |
| A1. Seules les protéines ont une dynamique conformationnelle. | FAUX |
| A2. Toutes les protéines interagissent avec un gène. | FAUX |
| A3. Toutes les méthodes d'étude des interactions protéine-protéine permettent de déterminer la cinétique d'association de ces protéines. | FAUX |
| A4. Certaines protéines interagissent avec un gène. | VRAI |
| A5. Les méthodes physiques d'étude des interactions protéine-protéine permettent de déterminer la constante de dissociation de ces protéines. | VRAI |
| A6. Le pseudo-temps est le positionnement d'une cellule le long de la trajectoire qui quantifie la progression d'un processus biologique. | VRAI |
| A7. Une technique utilisant une/des protéine(s) de fusion est rapide et simple à appliquer. | FAUX |
| A8. La structure des macromolécules est l'élément central qui contrôlent leurs interactions. | VRAI |
| A9. Les anticorps sont un outils important pour l'étude des interactions protéine-protéine. | VRAI |
| A10. Les interactions protéines connues sont en très grande majorité issues de données expérimentales (biochimiques, biophysiques, …). | FAUX |
| A11. La technique appelée double-hybrides est une technique à très haut débit. | VRAI |
| A12. Tout comme les alignements de séquences et les arbres phylogénétiques, il n'y a pas de réseau d'interactions « juste » ou faux ». | VRAI |
| A13. Certaines protéines interagissent avec plusieurs dizaines d'autres protéines. | VRAI |
| A14. Toutes les protéines interagissent avec au moins une autre protéine. | FAUX |
| A15. Plus le nombre de molécules d'un ligand se fixant sur les sites de fixation d'une protéine est élevé, plus la variation d'enthalpie de cette réaction est faible. | FAUX |
| A16. Plus KD est petite, plus l'affinité de liaison du ligand pour son site de fixation est grande. | VRAI |
| A17. La prédiction de trajectoire décrit l'évolution de chaque cellule en ordonnant ses états selon son processus de développement. | VRAI |
| Voir l'ensemble de ressources dédiés aux interactions entre macromolécules biologiques. | |
Exercice 10 a. Analyser le paragraphe "2.2. Protein Enrichment Analysis" et la Figure 2 de l'article Chiaradia et al. (2019) "Proteome Alterations in Equine Osteochondrotic Chondrocytes" Int. J. Mol. Sci. 20, 6179 b. Partie "Materials and Methods" => paragraphe "4.4. Protein Enrichment Analysis" : quelles sont les 2 bases de données et les 2 applications ("plugins") de Cytoscape utilisés pour analyser les réseaux regroupés sur la base de la fonction des protéines dérégulées dans OC ? Réponse : ClueGO permet de visualiser les termes biologiques non redondants pour de grands groupes de gènes dans un réseau fonctionnellement regroupé.
CluePedia permet de rechercher de nouveaux marqueurs potentiellement associés à des voies ("pathways").
c. Quelles informations ontologiques et quelles ressources ont permis d'enrichir les réseaux ? Réponse : les processus biologiques, les fonctions moléculaires, le composant cellulaire, la base de données Reactome, les annotations de KEGG et de WikiPathways. d. Quels sont les groupes fonctionnels les plus importants ? Réponse : la glycolyse et la gluconéogenèse, le développement de la plaque de croissance et du cartilage, la régulation positive de l'import de protéines, l'activité du médiateur de l'adhésion cellule-cellule et le nucléoïde mitochondrial. |
| Liens Internet et références bibliographiques | |
Cours en ligne "Protein-protein interactions" Pathway Figure OCR : extraction d'informations publiées dans la littérature. Pathway Commons Cytoscape User Manual Figures d'articles scientifiques créées avec Cytoscape |
|
3D SARS-CoV-2-Human Interactome Browser SARS-CoV-2 Interactome 3D |
|
Chatr-aryamontri et al. (2007) "MINT: the Molecular INTeraction database" Nucleic Acids Res. 35, D572 - D574 Pavlopoulos et al. (2011) "Using graph theory to analyze biological networks" BioData Min. 4, 10 Szklarczyk et al. (2011) "The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored" Nucleic Acids Res. 39, D561 - D568 Kerrien et al. (2012) "The IntAct molecular interaction database in 2012" Nucleic Acids Res. 04, D841- D846 |
|
|
Fernandes et al. (2016) "Systematic analysis of the gerontome reveals links between aging and age-related diseases" Hum. Mol. Genet. 25, 4804 - 4818 Treutlein et al. (2016) "Dissecting direct reprogramming from fibroblast to neuron using single-cell RNA-seq" Nature 534, 391 - 395 Hu et al. (2017) "Functional Analysis of Human Hub Proteins and Their Interactors Involved in the Intrinsic Disorder-Enriched Interactions" Int. J. Mol. Sci. 18, 2761 |
|
|
Noor et al. (2019) "Biological insights through omics data integration" Curr. Opin. Sys. Biol. 15, 39 - 47 Ivarsson & Jemth (2019) "Affinity and specificity of motif-based protein-protein interactions" Curr. Opin. Struct. Biol. 54, 26 - 33 Cannoodt R. (2019) "Modelling single-cell dynamics with trajectories and gene regulatory networks" PhD thesis - Université de Gand Saelens et al. (2019) "A comparison of single-cell trajectory inference methods" Nat. Biotechnol. 37, 547 - 554 |
|
|
Gordon et al. (2020) "A SARS-CoV-2 protein interaction map reveals targets for drug repurposing" Nature 583, 459 - 468 Gogl et al. (2020) "Dual Specificity PDZ- and 14-3-3-Binding Motifs: A Structural and Interactomics Study" Structure 28, 747 - 759 Bajpai et al. (2020) "Systematic comparison of the protein-protein interaction databases from a user's perspective" J. Biomed. Inform. 103, 103380 |
|
|
Karatzas et al. (2022) "The network makeup artist (NORMA-2.0): distinguishing annotated groups in a network using innovative layout strategies" Bioinform. Adv. 2, vbac036 Kim et al. (2023) "A proteome-scale map of the SARS-CoV-2-human contactome" Nat. Biotechnol. 41, 140 - 149 Szklarczyk et al. (2023) "The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest Nucleic Acids Res. 51, D638 - D646 |
|
|
Kurbatov et al. (2023) "The Knowns and Unknowns in Protein–Metabolite Interactions" Int. J. Mol. Sci. 24, 4155 Teulière et al. (2023) "Interactomics: Dozens of Viruses, Co-evolving With Humans, Including the Influenza A Virus, may Actively Distort Human Aging" Mol. Biol. Evol. 40, msad012 Michaelis et al. (2023) "The social and structural architecture of the yeast protein interactome" Nature 624, 192 - 200 |
|
Lim et al. (2024) "Advances in single-cell omics and multiomics for high-resolution molecular profiling" Exp. Molec. Med. Cui et al. (2024) "scGPT: toward building a foundation model for single-cell multi-omics using generative AI" Nat. Methods 21, 1470 - 1480 You et al. (2024) "Systematic comparison of sequencing-based spatial transcriptomic methods" Nat. Methods 21, 1743 - 1754 Wu et al. (2024) "Simultaneous single-cell three-dimensional genome and gene expression profiling uncovers dynamic enhancer connectivity underlying olfactory receptor choice" Nat. Methods 21, 974 - 982 |
|