| Lexique
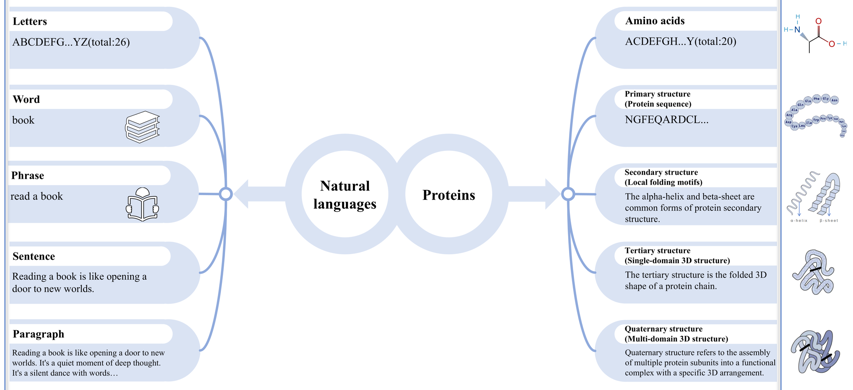
Dans ce cours, une phrase ou une séquence ont le sens "phrase d'un texte littéraire ou séquence de mots" ou le sens "séquences d'acides aminés".
|
| Les grands modèles de langage |
L'apprentissage profond et les grands modèles de langage ("Large Language Models" - LLM) sont des modèles d'apprentissage automatique conçus pour des tâches de traitement du langage naturel. |
| Les jetons ou "token" |
Les jetons ("tokens") ou unités de reconnaissance ("units of recognition") correspondent à des découpages variables de la phrase ou de la séquence en entrée.
Les jetons (un mot, un sous-mot ou un caractère, selon le modèle) sont donc les unités de base des LLM. |
| Le découpage des phrases ou séquences en entrée en jetons : la "tokenization" |
Le choix de la méthode de tokenisation est capital pour la performance du LLM.
Exemples de découpage en jetons (ou "tokenization") :
- Une séquence d'acides aminés découpée caractère par caractère (exemple : le code à 1 lettre des acides aminés).
- Un texte découpé par morceaux de mots (un même mot est découpé en plusieurs unités, pas nécessairement syllabiques) : le mot
"soleil" peut, par exemple, être découpé en "so/le/il".
- Un texte découpé mot par mot ou phrase par phrase.
Voir une application en ligne de tokenization. |
Exemples de découpage en jetons de différents types de données :
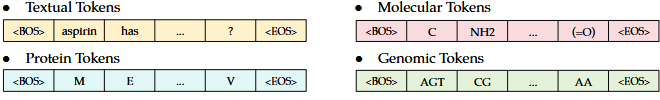
Source : Zhang et al. (2024)
|
| Exemple d'algorithme de "tokenization" |
L'un des premiers algorithmes, BPE ("Byte Pair Encoding" - codage des paires d'octets) identifie les paires de caractères adjacents les plus fréquentes dans une séquence et les remplace toutes par un caractère absent de la séquence entrée.
Le processus est répété jusqu'à ce qu'aucune compression supplémentaire ne soit possible (soit il n'y a plus de paires de caractères fréquentes, soit il n'y a plus de caractères non utilisés pour représenter ces paires).
- Exemple de séquence entrée : EY EY L EY L N
- Compression de la paire EY avec un caractère X non utilisé : X XL XL N
- Compression de la paire XL avec un caractère Z non utilisé : XZZD
BPE est utilisé pour le pré-entraînement de nombreux modèles de type Transformer (exemples : GPT2 et BART).
|
| Les n-grammes |
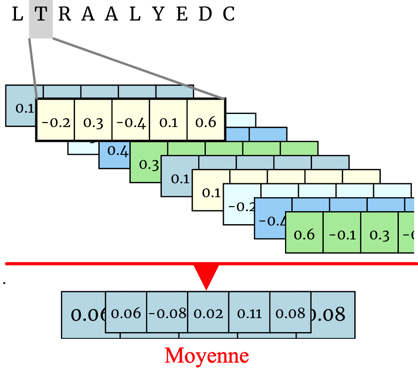
Un n-gramme ("n-gram") est une sous-séquence de n éléments adjacents dans un ordre particulier de la phrase ou de la séquence entrée.
Elle peut, par exemple, être une sous-séquence de n acides aminés consécutifs d'une séquence protéique.
Exemples de 3-grammes (trigramme) de la séquence "YFDKCWHL" : "YFD", "FDK", "DKC", "KCW"....
|
| Segmentation des séquences en acides aminés |
Pour une valeur n donnée, le nombre total de n-grammes possibles est 20n puisqu'il y a 20 acides aminés.
Pour n = 1 (appelé unigramme), un résidu d'acide aminé est donc traité comme un mot et une séquence protéique comme une phrase.
|
| Encodage "one-hot" |
En traitement du langage naturel :
- Un vecteur "one-hot" est une matrice de (1 × N) vecteurs utilisée pour distinguer chaque mot d'un vocabulaire de tous les autres mots de ce vocabulaire.
- Le vecteur est constitué de 0 dans toutes les cases, à l'exception d'un seul 1 dans une case unique pour identifier le mot considéré.
- Un groupe de bits "one-hot" est un groupe au sein duquel les combinaisons de valeurs ne possèdent qu'un bit à 1 et tous les autres à 0.
Exemples du codage des chiffres : 0 (décimal) = 00000001 ("one-hot"); 1 (décimal) = 00000010; 2 (décimal) = 00000100; ... ; 7 (décimal) = 10000000
|
| Inconvénient de l'encodage "one-hot" |
Soit un corpus de 1000 mots : l'encodage "one-hot" de chaque mot génère un vecteur de dimension 1000 (uniquement des 0 sauf à la position unique correspondant au mot, où la valeur = 1).
Cet encodage présente donc l'inconvénient d'une dimensionnalité très élevée si le corpus est étendu. |
| Encodeur - décodeur |
Un autoencodeur est un réseau de neurones pour l'apprentissage automatique non supervisé.
Un autoencodeur est composé d'un encodeur qui encode une phrase d'entrée et d'un décodeur qui reconstruit cette phrase à partir du code.
- Le modèle reconstruit les entrées en prédisant une valeur cible V' la plus proche de la valeur de l'entrée V.
- Une fonction de coût essaye de minimiser la distance entre V et V'.
- L'erreur est rétropropagée de la dernière à la première couche afin d'ajuster les poids de chaque neurone : ceux qui contribuent le plus à l'erreur sont davantage modifiés.
- Cette démarche permet d'entraîner le réseau de neurones du modèle d'apprentissage.
Les modèles [encodeurs-décodeurs] ne sont pas tous des auto-encodeurs. |
| Technique dite d'attention |
Processus qui permet aux modèles de traitement du langage naturel de pondérer les données d'entrée pour accorder plus d'importance aux données les plus pertinentes tout en ignorant d'autres.
|
| Complétion & inférence |
Dans la génération d'une réponse (une complétion), le modèle procède jeton après jeton.
Etant donnée une ébauche de réponse inachevée (une phrase tronquée par exemple), le modèle lui adjoint le jeton le plus probable (la probabilité de ce jeton étant calculée à partir des données d'entraînement) : c'est l'inférence. |
Principaux types de codage positionnel |
Le codage de position absolue est très utilisé en raison de sa simplicité et de son efficacité de calcul.
- Cependant, dans le cas d'une séquence polypeptidique, ce type de codage nécessite d'encoder chaque position d'acide aminé, ce qui limite la capacité d'un modèle de traitement du langage protéique (pLM) à gérer des séquences protéiques de longueurs variables et sa capacité d'extrapolation.
- Le codage de position absolue appris conduit souvent à de meilleures performances en aval d'un pLM (exemple : ESM-1b).
Le codage de position relative est insensible à la longueur totale de la séquence, ce qui le rend plus adapté à la gestion de séquences de longueurs arbitraires et à la capture d'informations de structure.
|
| Voir un ensemble de vidéos très explicatives de l'ensemble de ces notions (modèle Transformer). |
| Les représentations distribuées de mots |
En traitement du langage naturel, elles désignent une manière de représenter des mots ou des phrases sous forme de vecteurs continus dans un espace vectoriel de grande dimension.
Elles aident les algorithmes d'apprentissage en regroupant les mots similaires.
|
| Notion de contexte |
Un vecteur d'intégration ("plongement") contextualisé pour un jeton ("token") inclue son contexte environnant : en conséquence, 2 jetons identiques n'ont pas forcément le même vecteur d'intégration contextualisé. |
| Technique "word2vec" |
Développée en 2013 par des chercheurs de Google : elle utilise des réseaux de neurones superficiels pour associer des mots uniques à des représentations distribuées de mots. |
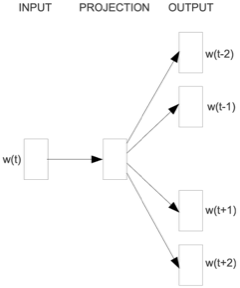
| "Skip-gram Word2Vec" |
C'est une architecture qui permet de calculer des représentations de mots ("word embeddings").
Au lieu d'utiliser les mots environnants pour prédire le mot central, "Skip-gram" utilise le mot central pour prédire les mots environnants.
|
| Sémantique |
"Etude du sens des unités linguistiques et de leurs combinaisons" (déf. Larousse).
Les termes similaires sur le plan sémantique sont plus susceptibles d'avoir des plongements ("word embeddings") proches. |
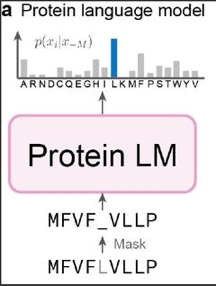
| pLM |
Les modèles de traitement du langage protéique ("protein Language Models"). |
| Classement de certains LLM selon leurs types de tâches |
| Architecture |
Exemples |
Caractéristiques principales |
Les modèles d'auto-encodage
Encodeur seul |
BERT ("Bidirectional Encoder Representations from Transformers" (Google 2018), XLNet, ELECTRA. |
- Ils saisissent le contexte des mots d'une phrase en prédisant les mots masqués.
- Performants dans la compréhension approfondie du contexte et de la sémantique.
|
Les modèles autorégressifs
Décodeur seul |
La famille des modèles GPT (GPT-3.5 à GPT5 - "Generative Pretrained Transformer", OpenAI). |
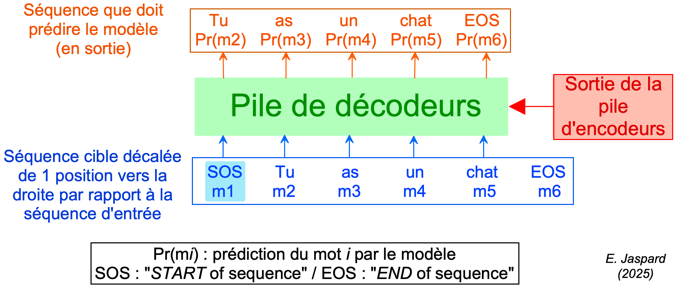
- Modèle unidirectionnel qui prédit le jeton ("mot") suivant d'une séquence en fonction de tous les jetons qui précèdent (prédiction de gauche à droite) en conservant l'ordre d'origine des jetons.
- Ces modèles sont performants dans la génération de texte.
- Ces modèles sont performants dans la génération de texte.
|
Les modèles séquence à séquence (Seq2Seq)
Encodeur-décodeur |
T5 ("Text-To-Text Transfer Transformer"), BART. |
- Conçus pour les tâches où l'entrée et la sortie sont des séquences.
- Génération de séquences de sortie à partir de séquences d'entrée masquées.
- Ces modèles sont performants dans la transformation d'un type de texte en un autre (traduction de langues, conversion de longs textes en résumés).
|
c. Nombre croissant des modèles d'apprentissage automatique appliqués à la biologie
On assiste à une augmentation spectaculaire des publications de ces modèles ayant pour but de décrypter des processus biologiques (généraux ou spécifiques).
En voici quelques exemples :
|
| Transcription et régulation de processus ARN-dépendants |
Annotation de la structure topologique (données issues de la technique d'analyse du génome Hi-C) pour capturer la conformation de la chromatine.
Prédiction (et amélioration de l'interprétabilité de cette prédiction) des sites de liaison des facteurs de transcription.
Modélisation de l'élongation de la transcription.
Prédiction des événements d'épissage alternatif sans données de référence.
Prédiction de la localisation multicompartimentale des ARN et de la localisation subcellulaire des ARNm.
Identification des sites de liaison [ARN circulaire - protéine de liaison à l'ARN] ou [lncRNA - protéine de liaison à l'ARN].
Prédiction des interactions [miRNA - lncRNA] de végétaux.
Découverte de régions pré-miRNA dans les génomes de végétaux.
|
| Système CRISPR/Cas9 |
Prédiction de l'activité cible de l'ARN guide unique.
Utilisation des informations de séquence pour prédire l'activité hors cible de CRISPR-Cas9.
Prédiction de la spécificité hors cible et de l'aptitude cellulaire du système CRISPR-Cas.
Prédiction de l'efficacité et de la spécificité du clivage de l'ARN guide unique.
|
| Protéines et ligands |
Identification de la localisation sub-mitochondriale des protéines mitochondriales.
Détection des séquences signal dans les peptides d'adressage.
Prédiction de l'effet des variants sur les protéines multidomaines.
Prédiction des résidus d'acides aminés impliqués dans la fixation de diverses classes de ligands.
|
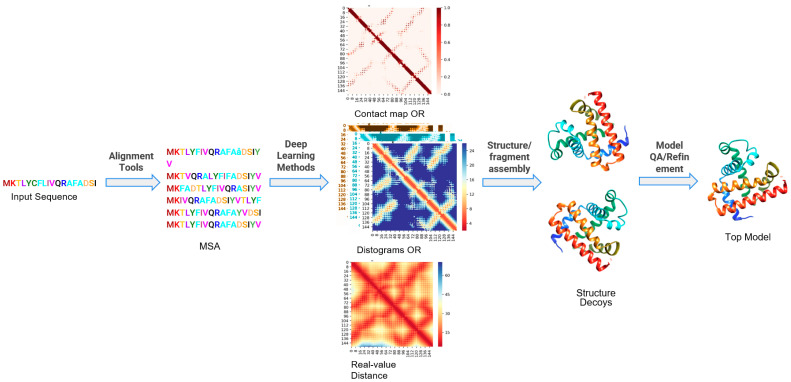
| Structure des protéines |
Prédiction du repliement des protéines, de la topologie des protéines transmembranaires α-hélicoïdales, du nombre de sous-unités dans les complexes protéiques homo-oligomériques, de la structure quaternaire des protéines, ...
|
| Single-cell |
Analyse de données RNAseq unicellulaires.
Déduction de réseaux de régulation des gènes à partir de données transcriptomiques unicellulaires.
Identification du type cellulaire à partir de données RNAseq unicellulaires.
|
| Maladies et cancers |
Prédiction de la corrélation [lncRNA - maladie], de la corrélation [miRNA - maladie].
Méthode d'intégration multi-omique pour la classification des données biomédicales.
Prédiction des interactions médicamenteuses.
Prédiction des régulateurs de la méthylation de l'ADN dans le cerveau humain et analyse de la génétique des troubles psychiatriques.
Réponses médicamenteuses dans certains cancers.
Prédiction de la sensibilité aux anticancéreux.
Prédiction et classification des sous-types de cancer du sein.
|
| Géographie |
Utilisation des terres et classification de l'occupation des sols.
Transfert intercontinental d'un modèle de cartographie du riz.
|
| Source : Choi & Lee (2023). |
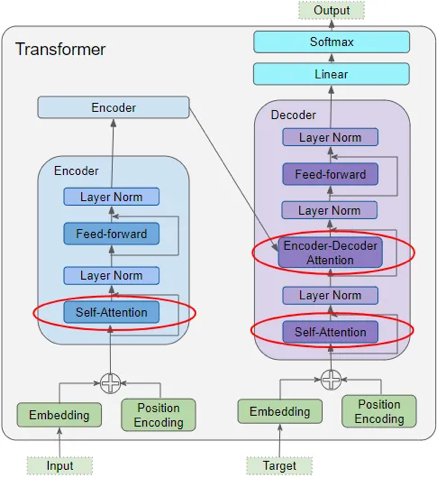
d. Le mécanisme d'attention du modèle Transformer
α. Les matrices Wq, Wk et Wv
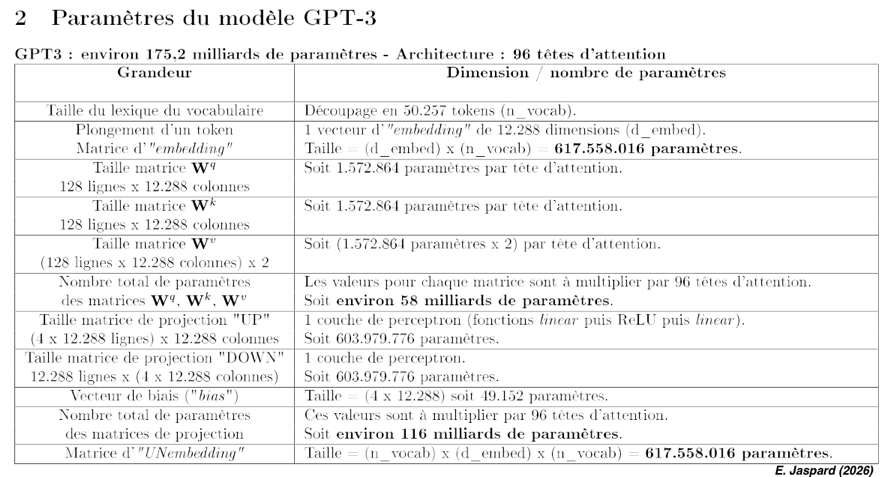
Chaque couche d'attention est caractérisée par 3 matrices de poids apprises par le transformeur : le poids de la requête ("Query" - Q), le poids de la clé ("Key" - K) et le poids de la valeur ("Value" - V) (figure ci-dessous).
Les matrices Wq, Wk et Wv permettent au modèle de se concentrer sur certains aspects de la séquence d'entrée afin d'en discerner les informations essentielles pour son encodage. |
| La matrice Wq ou matrice de requête |
La matrice Wk ou matrice de clé |
La matrice Wv ou matrice de valeur |
Elle représente l'information traitée du mot courant.
Elle facilite le processus de notation et permet d'évaluer la pertinence des autres mots par rapport au mot courant. |
Elle représente l'information traitée de tous les mots de la phrase, y compris le mot courant.
Elle permet de calculer un score qui représente la relation entre les différents mots de la phrase. |
Elle représente l'information brute de tous les mots de la phrase, y compris le mot courant.
Une fois les scores calculés entre les différents mots de la phrase, ces scores pondèrent la matrice de valeurs qui fournit une représentation agrégée des mots dans leur contexte. |
| Au début de l'apprentissage, ces matrices de pondération sont initialisées avec des valeurs aléatoires. |
β. Les vecteurs Q, K et V
Le vecteur d'intégration (représentation - "embedding") de chaque mot de la phrase d'entrée est multiplié par chacune de ces 3 matrices apprises afin de générer 3 vecteurs: Q, K et V.
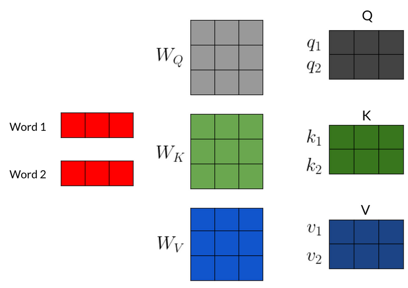
Source : Coursera |
| Le vecteur Q ou vecteur de requête |
Le vecteur K ou vecteur de clé |
Le mécanisme d'auto-attention calcule un score d'attention d'une paire de vecteurs [requête-clé].
Ce score est le produit scalaire des vecteurs Q et K. |
Le vecteur V ou vecteur de valeur |
| C'est le mot (ou "token") courant dont le modèle calcule la représentation (intégration - "embedding"). |
Ce sont les éléments auxquels est comparée la requête pour déterminer les poids (pondérations). |
- Les scores sont multipliés par la matrice Wv.
- Le vecteur de valeur est donc l'élément pondéré par le score d'attention.
|
Le modèle génère ainsi un encodage du contexte de chaque mot (ou "token") de la séquence analysée : le contexte est l'ensemble des mots, le mot considéré inclu.
En d'autres termes, chaque mot d'une séquence "communique" avec tous les autres mots, y compris lui-même.
Vidéos très pédagogues sur le mécanisme d'attention :
|
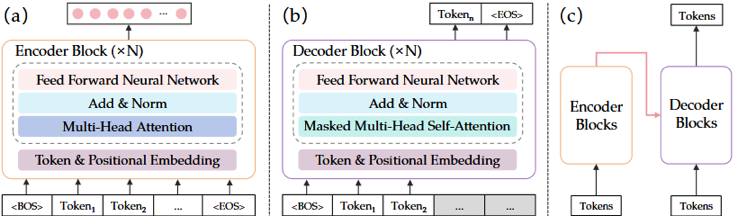
e. Les couches d'attention du modèle transformer
L'encodage du contexte de chaque mot (ou "token") constitue l'entrée de la couche suivante du modèle. |
| Couche d'auto-attention de l'encodeur ("Encoder Self-Attention layer") |
Modèle "Transformer" |
Couche d'auto-attention du décodeur ("Decoder Self-Attention") |
|
La représentation codée de chaque mot de la séquence d'entrée ("Input Embedding" - figure ci-contre) est transmise aux 3 matrices de poids d'entrée de la couche d'auto-attention du 1er encodeur.
Cette couche génère une nouvelle représentation codée de chaque mot de la séquence d'entrée en y intégrant le score d'attention associé à chaque mot.
Le traitement par plusieurs couches d'auto-attention permet d'ajouter de multiples scores d'attention à la représentation de chaque mot.
Cette représentation améliorée est transmise aux vecteurs requête et clé qui sont traités par la couche d'attention du 1er décodeur ("Encoder-Decoder Attention"). |
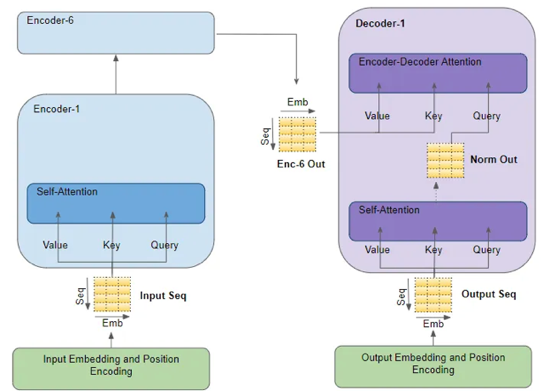
Source : "Transformers Explained Visually" |
La représentation contextuelle codée de chaque mot de la séquence cible ("Output Embedding") est transmise aux 3 matrices de poids de la couche d'auto-attention du 1er décodeur ("Self-Attention").
Cette couche génère une nouvelle représentation codée de chaque mot de la phrase cible en y intégrant les scores d'attention de chaque mot.
Après avoir traversé la couche de normalisation ("Norm Out"), cette représentation améliorée est transmise au vecteur requête de la couche d'attention ("Encoder-Decoder Attention") du 1er décodeur. |
La normalisation a pour but de modifier les données afin que leur moyenne = 0 et leur écart-type = 1 pour obtenir une distribution standard des données.
Voir la fonction "Normalize()" du module "transforms" de la bibliothèque PyTorch.
Formule du calcul d'attention :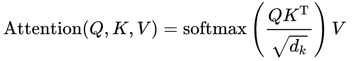 |
Les modèles de transformeurs (et les modèles d'apprentissage profond de manière générale), sont des objets complexes du point de vue mathématique et informatique.
Voir par exemple : "The Transformer Model in Equations : precise mathematical definition of the transformer model"
Thickstun J. |
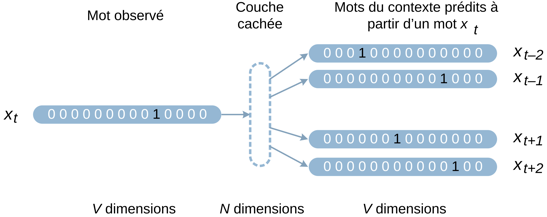
d. Illustration du plongement ou intégration ("embedding")
Le plongement ("embedding") encode les informations sémantiques (voir le lexique) significatives d'une phrase ou d'un mot sous la forme d'un vecteur de nombres réels (avec une table de correspondance, "lookup table") dans un espace vectoriel pouvant compter des milliers de dimensions.
- Considérons les intégrations (plongements) à 3 dimensions des mots similaires "apple" = [1,40, 0,02, -0,36] et "orange" = [1,52, 0,16, -0,11]. Les deux intégrations ont une valeur positive en 1ère position.
- Considérons les intégrations des mots similaires "apple" = [2,24, -0,03, -2,18] et "microsoft" = [-0,62, -0,38, -1,53]. Les deux ont une valeur négative en 3è position.
Par ailleurs, la manipulation (opérations arithmétiques, calculs matriciel, ...) de ces vecteurs permet de mieux évaluer la proximité des mots encodés.
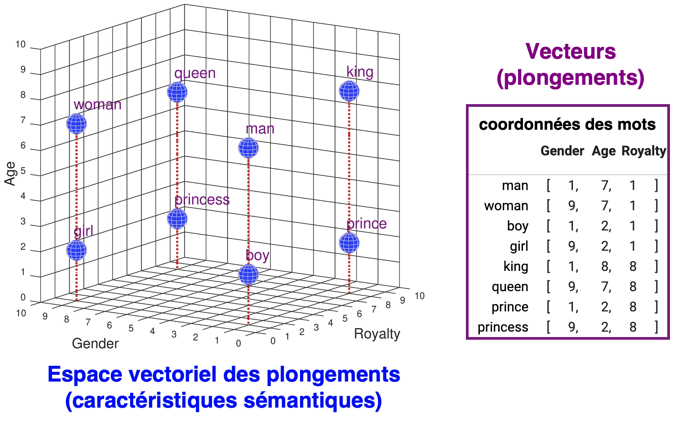
Source : "Word Embedding Demo : Tutorial"
Le plongement (ou représentation) sous forme de vecteurs permet un très grand nombre de calculs et d'opérations.
Par exemple, l'analogie par arithmétique vectorielle (figure ci-dessous) : le mot "man" est à "king" ce que le mot "woman" est à "king – man + woman" = "queen".
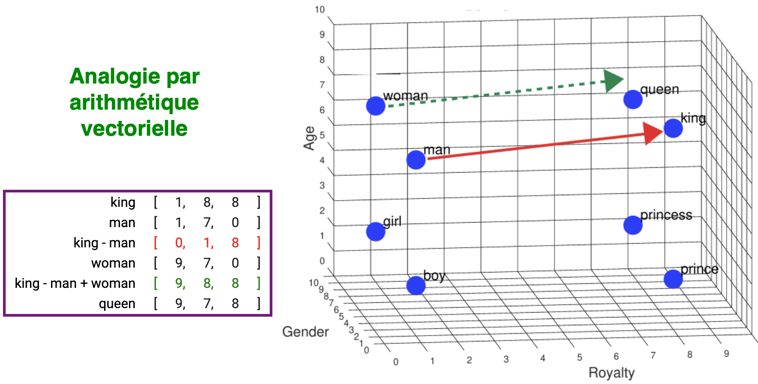
Source : "Word Embedding Demo : Tutorial"
|
| c. Jeux d'entraînement des pLM |
| SwissProtCLAP |
Jeu de données qui comprend 441.000 paires [texte - séquence protéique].
Il est construit à partir des données de la base de données SwissProt. |
| ProteinDT |
cadre de conception de protéines guidé par le texte, entraîné avec SwissProtCLAP. |
| ProtDescribe |
Jeu de données qui comprend 553.052 paires [séquences protéiques alignées - descriptions de leurs propriétés] (construit à partir des données de la base de données SwissProt). |
| UniProtQA |
Jeu de données dérivé d'UniProt pour l'entraînement du pLM "OpenBioMed".
Il comprend des protéines et des requêtes textuelles qui englobent 4 propriétés de ces protéines (caractéristiques fonctionnelles, nomenclature officielle, familles de protéines, localisation subcellulaire). |
| InstructProtein |
Jeu de données pour l'entraînement du pLM "OntoProtein" pour la génération bidirectionnelle entre le langage humain et le langage protéique. |
| ProteinKG25 |
Jeu de données de graphes de connaissances ("Knowledge Graph" - KG) à grande échelle.
ProteinKG25 contient ≈ 5 millions de corrélations [protéines - termes GO] et [termes GO - termes GO] et 31 relations. |
| ProteinLMBench |
1er jeu de référence pour l'évaluation des capacités de compréhension des pLM, notamment 944 questions à choix multiples soigneusement sélectionnées couvrant un large éventail de tâches liées aux protéines (voir l'article). |
| d. Exemples d'application des pLM en biologie |
| Génotype - phénotype |
Prédiction des conséquences phénotypiques à partir des millions d'effets possibles des variants faux-sens dans le génome humain. |
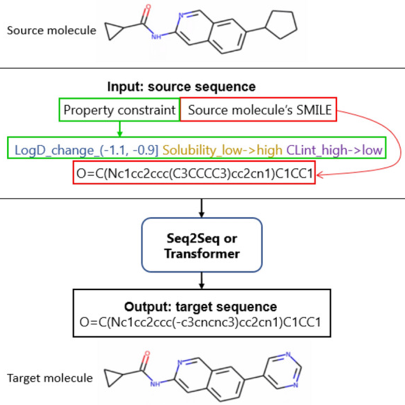
| Nouveaux médicaments |
Modèles de langage chimique à partir des bibliothèques de composés chimiques pour prédire des petites molécules médicaments capables de cibler des protéines impliquées dans des maladies. |
| Structure des protéines |
Prédiction de la fonction et de la structure des protéines. |
| Nouvelles protéines |
Génération de nouvelles protéines artificielles. |
| Lutte antivirale |
Guider l'évolution en laboratoire des variants d'anticorps neutralisant des virus (exemples : Ebola, SARS-CoV-2, ...). |
| Antibiorésistance |
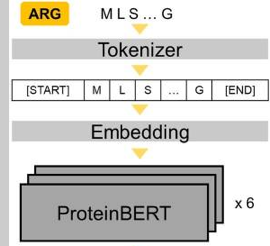
ARG-BERT : pLM qui utilise l'architecture de ProteinBERT pour la prédiction des mécanismes de résistance aux antibiotiques ("Antibiotic Resistance Genes" - ARG). |
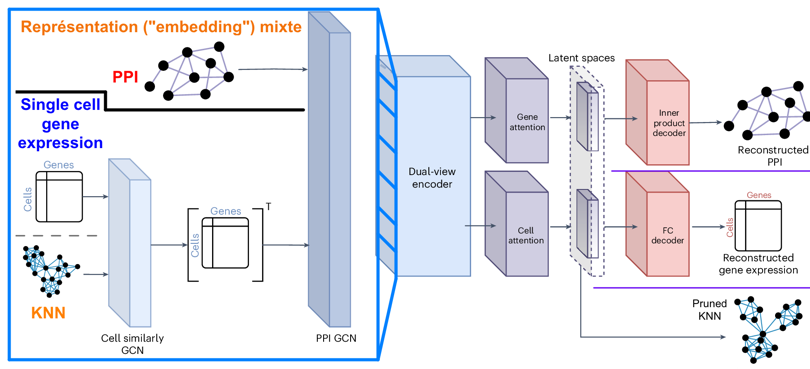
| Single-cell |
Transformeur scBERT ("single cell BERT") pour annoter avec précision les types de cellules à partir de données "single cell" - scRNAseq. |
| Réseaux de gènes |
Prédiction de la dynamique des réseaux de gènes pour la découverte de régulateurs clés de réseaux et de cibles thérapeutiques. |
| Ontologie |
GOProFormer : méthode de transformation multimodale pour la prédiction de l'ontologie de la fonction des protéines (annotation). |
| Evolution des protéines |
EVOLVEpro : méthode d'apprentissage permettant d'améliorer in silico l'évolution dirigée des protéines ("directed evolution of proteins") grâce à une combinaison de pLM et de prédicteurs de la fonction des protéines. |
| Anticorps |
AntiBERTy : pLM spécifique des anticorps, pré-entraîné avec 558 millions de séquences d'anticorps naturels. |
| Modifications post-traductionnelles |
PTG-PLM : pLM pour la prédiction des sites de glycosylation et de glycation.
pLMSNOSite et LMSuccSite : pLM pour la prédiction des sites de S-nitrosylation et de succinylation des protéines, respectivement |
| Voir un développement très détaillé : Zhang et al. (2024). |
| 8. Liens Internet et références bibliographiques |
|
pLM ("protein Language Model")
pLM de UNIPROT
ProtNLM ("Protein Natural Language Model") de UNIPROT
"BERT détaillé" |
pLM de UNIPROT
ProtNLM
Aller au site |
|
Transformeurs
Transformers
Transformers : architecture et code
Listes de transformeurs
Code du Transformer - "Notebook Pytorch" (transformer.ipynb)
|
Transformers 1
Transformers 2
Voir la liste
Notebook
|
|
Plongement - intégration - incorporation ("embedding")
The Illustrated Word2vec
Protvec : Amino Acid Embedding Representation for Machine Learning Features
bio_embeddings : python package & pipeline for embedding generation
bio-embeddings library 0.2.2 (Python)
Application en ligne : "Incorporations de mots à l'aide d'un modèle Keras simple" - TensorFlow (Google)
Exercices pratique "embedding"
|
Word2vec
Protvec
package
library
Application en ligne
Exercices en ligne
|
|
BioNeMo (NVIDIA)
Fonctions d'activation
Application en ligne "Tokenizers - BPE" (Python)
|
BioNeMo
Applet Java
Tokenizers
|
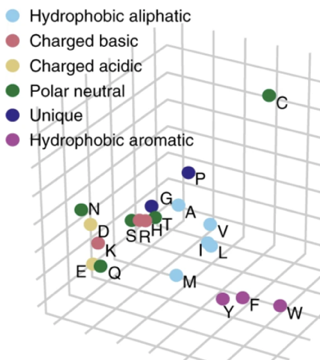
Lin et al. (2002) "Amino acid encoding schemes from protein structure alignments: multi-dimensional vectors to describe residue types" J. Theor. Biol. 216, 361 - 365
Mei et al. (2005) "A new set of amino acid descriptors and its application in peptide QSARs" Biopolymers 80, 775 - 786 |
Article
Article
|
Melvin et al. (2011) "Detecting remote evolutionary relationships among proteins by large-scale semantic embedding" PLoS Comput. Biol. 7, p.e1001047
Mikolov et al. (2013) "Efficient Estimation of Word Representations in Vector Space" arXiv:1301.3781
Asgari & Mofrad (2015) "Continuous Distributed Representation of Biological Sequences for Deep Proteomics and Genomics" PLoS One 10, e0141287 |
Article
Article
Article
|
|
Article du modèle"Transformer" : Vaswani et al. (2017) "Attention Is All You Need" arXiv:1706.03762
Extrait de l'article : "Nous proposons une nouvelle architecture de réseau simple, le Transformer, basée uniquement sur des mécanismes d'attention, se passant entièrement de récurrence et de convolutions."
"Transformer's Encoder-Decoder" : site très explicatif des notions liées au "Transformer" et à l'apprentissage (beaucoup de scripts Python)
|
Article
KiKaBeN
|
|
Krstovski & Blei (2018) "Equation Embeddings arXiv:1803.09123
F. Chollet (2019) "On the Measure of Intelligence" arXiv 1911.01547
ElAbd et al. (2020) "Amino acid encoding for deep learning applications" BMC Bioinformatics 21, 235
Raffel et al. (2020) "Exploring the Limits of Transfer Learning with a Unified Text-to-Text Transformer" J. Mach. Learn. Res 21, 1 - 67 |
Article
Article
Article
Article
|
|
Jumper et al. (2021) "Highly accurate protein structure prediction with AlphaFold" Nature 596, 583 - 589
Rives et al. (2021) "Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences" Proc. Natl. Acad. Sci. USA 118, e2016239118
Dallago et al. (2021) "Learned Embeddings from Deep Learning to Visualize and Predict Protein Sets" Curr. Protocols in Bioinformatics 10.1002/cpz1.113
He et al. (2021) "Molecular optimization by capturing chemist's intuition using deep neural networks" J. Cheminform. 13, 26 |
Article
Article
Article
Article
|
|
Baek et al. (2022) "Accurate prediction of protein structures and interactions using a 3-track neural network" Science 373, 871 - 876
Elnaggar et al. (2022) "ProtTrans: Toward Understanding the Language of Life Through Self-Supervised Learning" IEEE Trans. Pattern Anal. Mach. Intell. 44, 7112 - 7127
Pakhrin et al. (2022) "Deep Learning-Based Advances in Protein Structure Prediction" Int. J. Mol. Sci. 22, 5553
|
Article
Article
Article
|
|
Bandyopadhyay et al. (2022) "Interactive Visualizations of Word Embeddings for K-12 Students" EAAI-22
Ferruz et al. (2022) "ProtGPT2 is a deep unsupervised language model for protein design" Nat. Commun. 13, 4348
Brandes et al. (2022) "ProteinBERT: a universal deep-learning model of protein sequence and function" Bioinformatics 38, 2102 - 2110
|
Article
Article
Article
|
|
Kaminski et al. (2023) "pLM-BLAST: distant homology detection based on direct comparison of sequence representations from protein language models" Bioinformatics 39, btad579
Madani et al. (2023) "Large language models generate functional protein sequences across diverse families" Nat. Biotechnol. 41, 1099 - 1106
Pokharel et al. (2023) "Integrating Embeddings from Multiple Protein Language Models to Improve Protein O-GlcNAc Site Prediction" Int. J. Mol. Sci. 24, 16000
|
Article
Article
Article
|
|
Lin et al. (2023) "Evolutionary-scale prediction of atomic-level protein structure with a language model" Science 379, 1123 - 1130
Elnaggar et al. (2023) "Ankh : optimized protein language model unlocks general-purpose modelling" bioRxiv doi: 10.1101/2023.01.16.524265
Avraham et al. (2023) "Protein language models can capture protein quaternary state" BMC Bioinformatics 24, 433
Choi & Lee (2023) "Transformer Architecture and Attention Mechanisms in Genome Data Analysis: A Comprehensive Review" Biology (Basel) 12, 1033
|
Article
Article
Article
Article
|
Heinzinger et al. (2024) "Bilingual language model for protein sequence and structure" NAR Genom. Bioinform. 6, lqae150
Zhang et al. (2024) "Scientific Large Language Models: A Survey on Biological & Chemical Domains" arXiv:2401.14656 |
Article
Article
|
Hayes et al. (2025) "Simulating 500 million years of evolution with a language model" Science 387, 850 - 858
Wang et al. (2025) "A Comprehensive Review of Protein Language Models" arXiv, 2502.06881v1
Sheinin et al. (2025) "scNET: learning context-specific gene and cell embeddings by integrating single-cell gene expression data with protein–protein interactions" Nat. Methods 22, 708–716
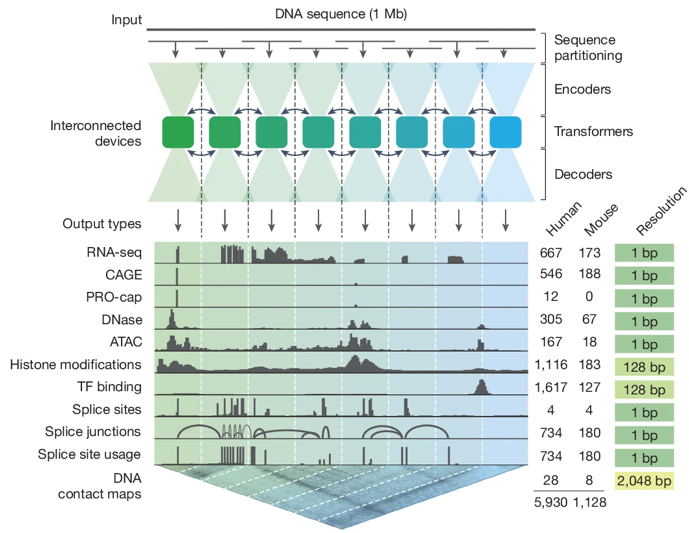
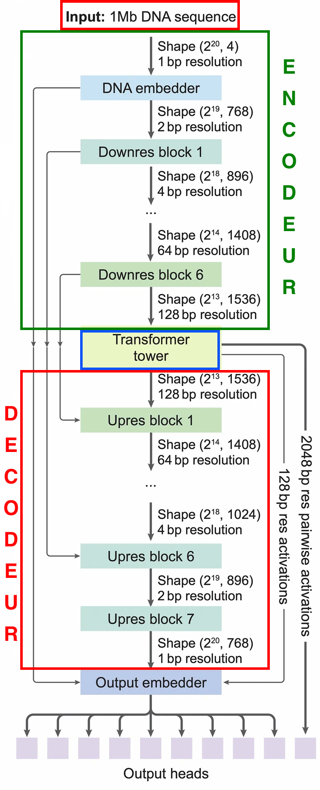
Garcia-Gonzalez & Gogolewski (2025) "AlphaGenome: a Swiss-army knife for exploring non-coding DNA" Trends Genet. 42, 4 - 6
A. Jolicoeur-Martineau (2025) "Less is More: Recursive Reasoning with Tiny Networks" arXiv:2510.04871v1 |
Article
Article
Article
Article
Article |
Wang et al. (2025) "Hierarchical Reasoning Model" arXiv 2506.21734v3
Siwek et al. (2025) "Sliding Window Interaction Grammar (SWING): a generalized interaction language model for peptide and protein interactions" Nat. Methods 22, 1707 - 1719
Chen et al. (2025) "Target sequence-conditioned design of peptide binders using masked language modeling" Nat. Biotechnol. doi: 10.1038/s41587-025-02761-2
Cherry & Qian (2025) "Supervised learning in DNA neural networks" Nature 645, 639 - 647
Olson et al. (2025) "NEAR: neural embeddings for amino acid relationships" Bioinformatics 41, i449–i457
Asim et al. (2025) "Protein Sequence Analysis landscape: A Systematic Review of Task Types, Databases, Datasets, Word Embeddings Methods, and Language Models" Database (Oxford) baaf027 |
Article
Article
Article
Article
Article
Article
|
Avsec et al. (2026) "Advancing regulatory variant effect prediction with AlphaGenome" Nature 649, 1206 - 1218 |
Article |
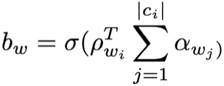
{kind=link}