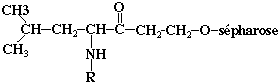| Purification et caractérisation de l'endoprotéase L de E. coli |
| Tweet |
|
|
1. Enoncé 2. Réponse partie A 3. Réponse partie B 4. Réponse partie C a. Détermination de la masse molaire d'une molécule par filtration sur gel |
c. L'électrophorèse sur gel bi-dimensionnel 5. Réponse partie D a. Rappels sur les hélices α 6. Liens Internet et références bibliographiques |
|
1. Enoncé L'endoprotéase L de Escherichia coli coupe les liaisons peptidiques entre les acides aminés Leu et X, X étant n'importe quel acide aminé. Partie A On teste plusieurs méthodes de purification de cette enzyme sur un extrait brut de E. coli. Les résultats sont les suivants : |
| Méthode | Extrait brut | Après purification | ||||
| volume (mL) | Q (mg) | AT (U) | volume (mL) | Q (mg) | AT (U) | |
| gradient de saccharose | 2 | 12 | 300 | 5,5 | 0,96 | 240 |
| chromatographie gel séphadex G200 | 10 | 60 | 1500 | 30 | 4,8 | 1200 |
| chromatographie gel DEAE-cellulose | 50 | 300 | 7500 | 8 | 7,5 | 7000 |
| précipitation sulfate ammonium (entre 60% et 80%) | 200 | 1200 | 30000 | 10 | 120 | 5000 |
| Q (mg) = quantité de protéines (mg) / AT (U) = Activité Totale (unités arbitraires) | ||||||
|
a. Rappeler le principe des
techniques utilisées. b. Calculer le rendement et le facteur de purification pour chaque technique utilisée. c. Quelles sont les deux méthodes les plus performantes et pourquoi ? Dans quel ordre les utiliser ? |
|
Partie B Pour purifier davantage l'enzyme, un gel d'affinité est synthétisé : une molécule Y est fixée de manière covalente sur de la sépharose et le gel Z est obtenu (structure ci-dessous).
Pourquoi ce gel permet-il de purifier davantage l'endoprotéase L ? Quel est le mode opératoire ? |
|
Partie C Une fois l'enzyme purifiée, elle est caractérisée par les techniques suivantes :
a. Rappeler le principe des techniques utilisées. Partie D Les polypeptides A et B ont une teneur en hélice α de 20 %. a. Rappeler les caractéristiques des hélices α. |
|
Centrifugation en gradient de densité La vitesse de sédimentation d'une particule (organite, macromolécule, ...) est fonction de la différence entre sa densité et celle du milieu ambiant. Les variations de densités sont obtenues en faisant varier la concentration d'un produit chimique qui doit être très solubles dans l'eau. Divers produits peuvent être utilisés pour faire ces gradients. |
| produit | densités | avantage | inconvénient |
| saccharose | 1,3 g/mL - 2,5 M | peu coûteux / électriquement neutre | problème de pression osmotique : à éviter pour l'isolement de cellules entières |
| chlorure de césium (CsCl) |
1,9 g/mL - 7,5 mol/L | densité très élevé trés adapté pour la séparation des acides nucléiques |
coût / sel |
| Metrizamide® (dérivé iodobenzamido du glucose) |
1,5 g/mL | très soluble et non chargé | coût / forte absorption dans l'UV / instabilité |
| Ficoll® (polymère de glucose) |
densités élevées
sans générer de fortes pressions osmotiques >adapté à la séparation de cellules entières |
coût / forte viscosité | |
| Percoll® (silice recouverte de polyvinylpyrolidone) |
1,3 g/mL | faibles pressions osmotiques
et faible viscosité adapté à la séparation de cellules entières |
coût / faibles densités atteintes |
|
La vitesse de sédimentation peut donc être modifiée en faisant varier de façon continue ou discontinue (par paliers) cette différence en créant un gradient de concentration du milieu ambiant.
Source : "Centrifugation en gradient de densité" |
|
Chromatographie d'exclusion ou filtration sur gel Le principe de la chromatographie d'exclusion (appelée aussi filtration sur gel ou tamisage moléculaire ou chromatographie de perméation) repose en première approximation sur la masse molaire des molécules à séparer. En toute rigueur, le paramètre physico-chimique qui intervient est la dimension des molécules, donc leur volume et donc plus précisément leur rayon de Stokes. Mais cette relation univoque [masse molaire - rayon de Stokes] n'est valable que pour des protéines dites "globulaires", c'est-à-dire assimilées à des sphères Différents paramètres influencent le volume des molécules et peuvent fausser la détermination de la masse molaire réelle par ce type de chromatographie :
La phase stationnaire est constituée de billes de polysaccharides (type "Sephadex™") gonflées dans le solvant utilisé pour l'élution :
|
|
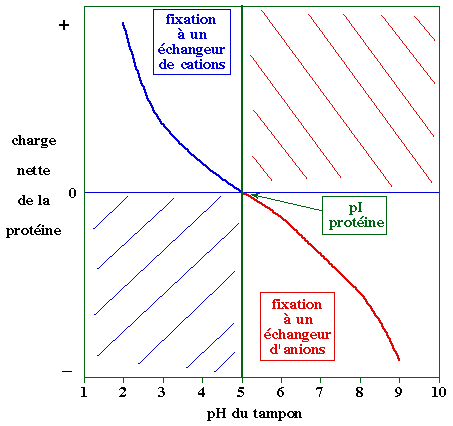
Chromatographie d'échange d'ions Les échangeurs d'ions sont des billes qui portent des groupements ionisables dont la charge est : |
| positive : résines échangeuses d'anions qui fixent des molécules chargées négativement : ---R+... M- |
négative : résines échangeuses de cations qui fixent des molécules chargées positivement : ---R-... M+ |
||
| nature de la fonction ionisable : ---R+ | nature de la fonction ionisable : ---R- | ||
| base forte | base faible | acide fort | acide faible |
| ammonium quaternaire :
---NR3+ exemple : triméthylammonium ---N(CH3)3+ |
forme protonnée
d'une amine I, II ou III : ---NHR2+ exemple : diéthylamino-éthylammonium 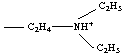 |
exemple : sulfonate ---SO3- |
exemple : carboxyméthyl ---O-CH2-CO2- |
|
Dans un premier temps, la résine est équilibrée dans un tampon dont le pH est tel que le groupement porté par l'échangeur d'ions soit ionisé :
Il y a deux manières d'éluer les molécules fixées sur la résine :
|
|
AT
étape n Rendement (%) = ---------------- AT étape 1 |
AT
étape n AS (U/mg) = ------------------------- Q protéines étape n |
AS
étape n FP = ---------------- AS étape 1 |
| AT : activité totale - AS : activité spécifique - FP : facteur de purification | ||
| Méthode | Extrait brut | Après purification | ||
| AS initiale (U/mg) | Rendement (%) | AS (U/mg) | FP | |
| gradient de saccharose | 25 | 80 | 250 | 10 |
| chromatographie gel séphadex G200 | 25 | 80 | 250 | 10 |
| chromatographie gel DEAE-cellulose | 25 | 93 | 933 | 37 |
| précipitation sulfate ammonium (entre 60% et 80%) | 25 | 17 | 42 | 1,7 |
|
Qu'il s'agisse du rendement et du facteur de purification, la méthode la plus performante dans l'exemple présent est la chromatographie d'échange d'ions DEAE-cellulose. Le gradient de saccharose et la chromatographie sur gel séphadex G200 donnent les mêmes valeurs : un bon rendement (bien qu'inférieur) et un facteur de purification nettement moindre. Le gradient de saccharose est le moins sélectif du fait de la taille des tubes et du principe même de la technique, surtout quand il s'agit d'un extrait brut. La chromatographie sur gel de filtration est une technique trés délicate : elle nécessite des colonnes particulières (trés longues et de section trés petite) afin d'augmenter le nombre de plateaux théoriques de fractionnement des protéines et elle doit être réalisée à un débit trés faible. Pour qu'une de chromatographie sur gel de filtration soit efficace, il faut que l'échantillon soit le plus concentré possible. Il en va de même pour le gradient de saccharose où l'on ne peut utiliser un grand volume d'échantillon. Après une chromatographie d'échange d'ions, il faut se débarasser du sel qui a servi à éluer la protéine. Pour celà, il faut dialyser les fractions qui contiennent la protéine éluée. La dialyse entraîne une dilution. Il faut alors concentrer l'échantillon (précipitation au sulfate d'ammonium / concentration sur membrane). Or la dialyse et la concentration entraînent de fortes pertes de protéines (exemple : le trés mauvais rendement de l'étape de précipitation au sulfate d'ammonium - tableau ci-dessus). En conséquence, il est préférable de faire d'abord le gradient de saccharose ou la chromatographie sur gel de filtration puis la chromatographie d'échange d'ions. |
|
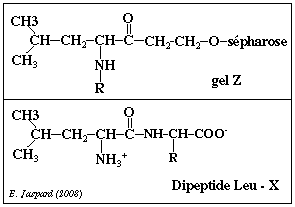
Le gel Z est un gel d'affinité. La molécule Y est un analogue structural de la leucine qui est le substrat de l'endoprotéase L de E. coli puisque cette dernière hydrolyse les liaisons peptidiques entre les acides aminés Leu et X.
La molécule Y (c'est-à-dire le bras du gel d'affinité) peut donc se fixer au niveau du site actif de de l'endoprotéase L sans pour autant être hydrolysé puisqu'il n'a pas une structure rigoureusement identique à celle du substrat. |
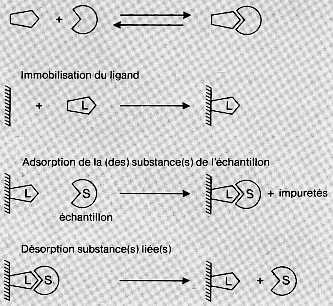
La chromatographie d'affinité est le type de chromatographie le plus sélectif, une protéine pouvant être purifiée d'un facteur 103 à 104 en une seule fois. Le principe de la chromatographie d'affinité repose sur la reconnaissance entre une molécule du mélange à séparer et une molécule greffée sur la résine, que l'on appelle le ligand, ces deux molécules interagissant de manière hautement spécifique :
En d'autres termes, on favorise l'équilibre molécule - [tierce molécule]. Cette tierce molécule peut-être :
|
Structure de gels d'affinité - Activation du gel pour le couplage du ligand - Bras espaceurs Les supports ou matrices utilisés pour la chromatographie d'affinité peuvent être des billes d'agarose (Sepharose), de polyacrylamide d'agarose, de verre poreux, de polyvinyle, de polyméthacrylate ... L'immobilisation du ligand sur la matrice nécessite qu'il soit préalablement activé, c'est-à-dire qu'il faut créer des sites réactionnels qui permettent d'établir des liaisons covalentes. Parmi les diverses méthodes d'activation, on peut citer :
La chromatographie d'affinité est la méthode la plus sélective. Cependant, c'est aussi la plus délicate à mettre en oeuvre car elle nécessite la mise au point des conditions expérimentales idoines pour obtenir un gel correctement activé :
|
|
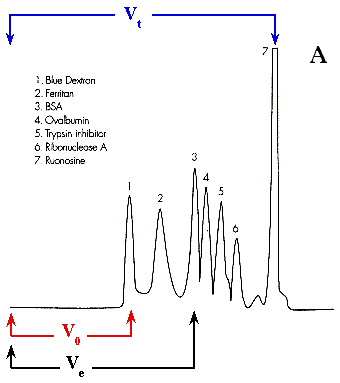
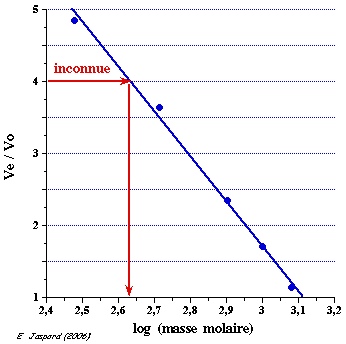
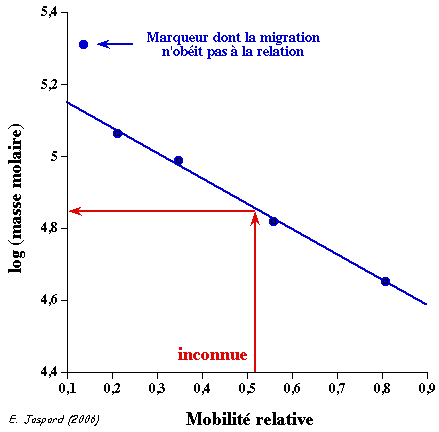
4. Réponse partie C a. Détermination de la masse molaire d'une molécule par filtration sur gel 1. Un mélange de molécules que l'on appelle standards, de masses molaires connues, est séparé par chromatographie sur gel de filtration (pics d'élution 1 à 7, figure ci-dessous).
Source : Chromatographie (Waters®) 2. Chaque molécule (caractérisée, par exemple, par un pic d'absorbance) a un volume d'élution Ve. 3. Le volume d'exclusion du gel (V0) est le volume d'élution d'une molécule non retardée, c'est-à-dire une molécule de taille supérieure au diamètre des pores des billes du gel et qui n'y diffuse donc pas. Bien souvent, on utilise le bleu dextran, polymère d'ose de masse molaire supérieure à 2 106 Da. 4. Le volume total du gel (Vt) est la somme du volume des billes et du volume externe aux billes. Il est donné par le volume d'élution d'une molécule qui diffuse totalement dans les billes (donc totalement retardée), ici la ruonosine. 5. A partir du profil d'élution des standards, on trace la la droite étalon : Ve / V0 = f log (masse molaire). 6. Dans les mêmes conditions de chromatographie que pour le mélange des standards, on détermine le volume d'élution d'une molécule inconnue. 7. On reporte la valeur du rapport Ve / V0 de la molécule inconnue sur la droite étalon et on détermine ainsi sa masse molaire.
|
|
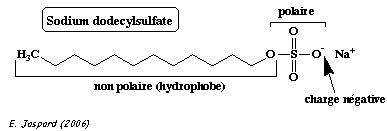
b. Gel d'électrophorèse des protéines en conditions dénaturantes La technique du gel d'électrophorèse en conditions dénaturantes ("sodium dodecyl sulfate polyacrylamide gel electrophoresis" ou SDS-PAGE) a été décrite par Ulrich Laemmli en 1970. C'est l'une des techniques (et donc l'un des articles) les plus cité(e)s dans la littérature scientifique : Laemmli, U. K. (1970) "Cleavage of structural proteins during the assembly of the head of bacteriophage T4" Nature 227, 680 - 685. On fait bouillir un mélange de protéines en présence :
En conséquence :
|
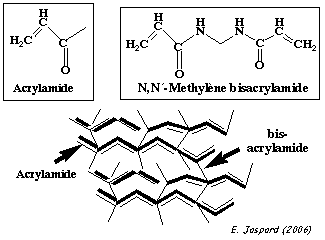
Les protéines de l'échantillon sont ensuite séparées par une électrophorèse sur gel de polyacrylamide en présence de SDS. C'est un gel réticulé, obtenu par polymérisation d'acrylamide qui forme des chaînes et de bis-acrylamide qui ponte les chaînes d'acrylamide.
La réaction de polymérisation est :
Plus le pourcentage d'acrylamide est élevé, plus la densité des chaînes est élevée et les mailles du réseau sont serrées. En conséquence :
|
|
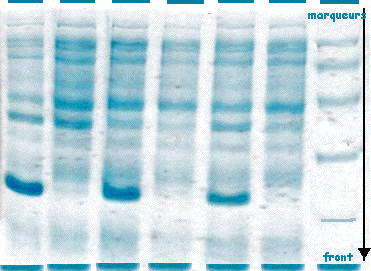
On obtient différentes bandes pour chaque piste de la figure ci-dessous (la flèche indique le sens de migration).
Source : E. Jaspard (2004) La masse molaire des protéines est déterminée à l'aide de marqueurs qui sont des protéines standards de masses molaires connues (piste de droite). Exemple de marqueurs : myosine (205 kDa), β galactosidase (116 kDa), phosphorylase b (97,4 kDa), albumine (66 kDa), ovalbumine (45 kDa), anhydrase carbonique (29 kDa). Détermination de la masse molaire d'une protéine
|
|
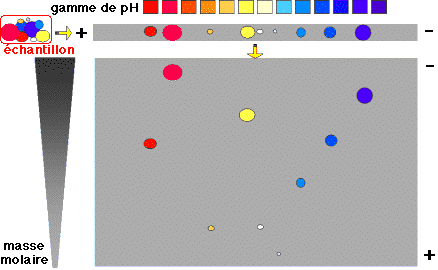
c. L'électrophorèse sur gel bi-dimensionnel (2DPAGE : "two-dimensional polyacrylamide gel electrophoresis") C'est la technique de base en protéomique. La stratégie la plus courante est de séparer dans un premier temps les protéines par électrophorèse bidimensionnelle en gel de polyacrylamide. Du fait de leur composition en acides aminés, les protéines diffèrent les unes des autres par :
Les protéines ont :
L'une des difficultés majeures de la séparation est de trouver des conditions d'électrophorèse qui couvrent de telles gammes de propriétés physico-chimiques. La séparation se fait en deux temps :
Source : Humboldt - Universität Berlin
Ces deux électrophorèses sont effectuées de manière perpendiculaire l'une par rapport à l'autre. Comme les paramètres de la séparation sont indépendants, cette technique est particulièrement résolutive : plusieurs centaines de chaînes polypeptidiques peuvent être séparées sous forme de taches de protéines ou spots sur un gel. Voir des exemples de gels bi-dimensionnels. Coloration des gels pour la révélation des protéines (100 ng à 0,1 ng de protéine par spot) :
Source : Sigma - Aldrich Les logiciels d'analyse d'image des gels repèrent les spots et calculent leur coordonnées et leur intensité. La sélection des spots à prélever est transmise au couteau à gel. Les morceaux de gels sont déposés dans les puits de plaques à 96 puits. |
|
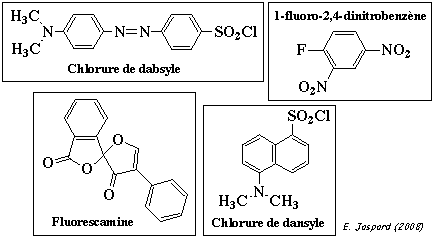
d. Identificaton de l'extrémité N-terminale et carte peptidique On détermine le résidu N-terminal d'une protéine en faisant agir, par exemple le chlorure de dansyle (DNS) (réactif fluorescent), le chlorure de dabsyle (réactif fluorescent), le 1-fluoro-2,4-dinitrobenzène (réactif de Sanger), l'isocyanate de methyle, la fluorescamine.
La carte peptidique est obtenue après digestion du polypeptide par une protéase (exemple : trypsine). La séparation des peptides issus de cette hydrolyse est effectuée par électrophorèse en conditions dénaturantes suivie d'une chromatographie de partage.
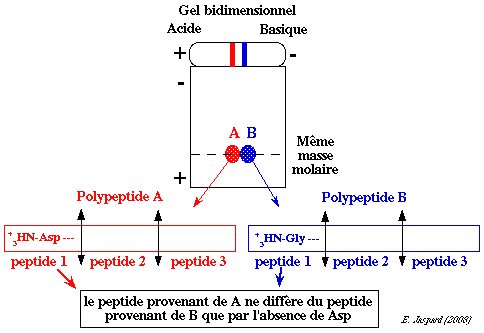
La technique la plus récente (appliquée en protéomique) est la spectromètrie de masse en tandem ou non. Comme l'indique la composition en acides aminés, le peptide provenant du polypeptide A ne diffère du peptide provenant du polypeptide B que par un Asp : il est donc plus négatif et plus hydrophile. En conséquence, les polypeptides A et B ne diffèrent que par leur extrémité N-terminale :
On peut supposer que les polypeptides A et B résultent de la duplication d'un gène ancestral unique puis divergence des 2 copies qui a entraîné la perte de l'acide aminé N-terminal Asp dans le polypeptide B. La masse molaire des polypeptides A et B est si proche que l'on observe qu'une bande sur gel d'électrophorèse en conditions dénaturantes. Structure de l'endoprotéase L de E. coli
L'endoprotéase L de E. coli est donc un tétramère qui pourraient être [2 chaînes polypeptide A + 2 chaînes polypeptide B]. |
|
5. Réponse partie D Deux hélices α adjacentes sont généralement arrangées de manière antiparallèle. Elles sont compactées par les liaisons hydrogène qui s'établissent entre les chaînes latérales des acides aminés. Ces paires (ou unités) sont souvent arrangées en faisceau à 4 hélices dans lequel les chaînes latérales des 4 hélices α sont empilées et forment un coeur hydrophobe au centre du faisceau. Les faisceaux à 4 hélices forment des domaines α dans les protéines. La myohémérythrine, le cytochrome b562, la petite protéine Rop qui se lie à l'ARN, sont des protéines de ce genre. b. Autres types d'hélices Hélice 310
Hélice π
|
| Hélices polyproline I et II | |
| Poly - Pro I | Poly - Pro II |
| liaisons peptidiques cis | liaisons peptidiques trans |
| hélice droite avec 3.3 résidus par tour | hélice gauche avec 3 résidus par tour |
| Φ = - 83° | Φ = - 78° |
c. Récapitulatif : voir la méthode du diagramme de Ramachandran pour une définition des angles. |
| type de structure | valeurs des angles (degrés) | nombre moyen de résidus par tour |
translation par résidu (Å) |
||
| Φ | Ψ | ω | |||
| hélice α | - 57 | - 47 | 180 | 3,6 | 1,5 |
| hélice 310 | - 49 | - 26 | 180 | 3,0 | 2,0 |
| hélice π | - 57 | - 70 | 180 | 4,4 | 1,15 |
| hélice polyproline I | - 83 | + 158 | 0 | 3,38 | 1,9 |
| hélice polyproline II | - 78 | + 149 | 180 | 3,0 | 3,12 |
| Source : base de données PROWL -> consulter l'item : "Residue hydrogen bonding" | |||||
Script Python pour représenter les diagrammes à partir d'un fichier de la PDB. |
|
d. Rappel sur le dichroïsme circulaire Le dichroïsme circulaire s'appuie sur la capacité des molécules qui ont une activité optique d'absorber différemment à droite et àgauche la lumière polarisée circulairement. L'activité optique est la propriété que possède une structure chirale d'interagir avec un rayonnement électromagnétique. Le dichroïsme circulaire est une technique qui permet d'analyser le contenu en structures secondaires des protéines ou des acides nucléiques. Pour déterminer la proportion de chaque type de structure secondaire, il faut effectuer une déconvolution en composantes élémentaires du spectre de dichroïsme avec des logiciels appropriés. Cette une technique non destructive qui permet d'étudier les changements de conformation des protéines dans différents environnements (pH, agents dénaturants, température). b. Lumière polarisée Certaines propriétés de la lumière sont celles d'une onde électromagnétique :
Dans la lumière naturelle, le vecteur E peut prendre toutes les directions perpendiculaires à la direction de propagation.
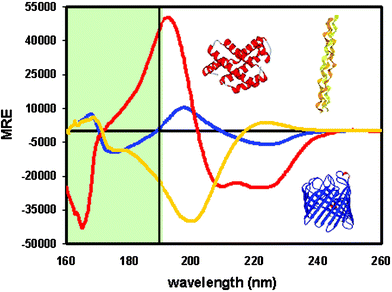
c. Spectres de dichroïsme circulaire La forme du spectre obtenu pour une protéine peut être déconvoluée (décomposée) en différentes composantes. Chaque composante donne une courbe spectrale caractéristique (figure ci-dessous) :
Source : Mile & Wallace (2006)
|
| 6. Liens Internet et références bibliographiques |
| Laemmli, U. K. (1970) "Cleavage of structural proteins during the assembly of the head of bacteriophage T4" Nature 227, 680 - 685 | |
|
Expasy - Swissprot : données d'électrophorèse sur gel bi-dimensionnel |
SWISS-2DPAGE |
|
Mile & Wallace (2006) "Synchrotron radiation circular dichroism spectroscopy of proteins and applications in structural and functional genomics" Chem. Soc. Rev. 35, 39 - 51 |
Article |
![]()