| Déshydrogénases et pli Rossmann / calmoduline et motif "EF-hand" / récepteur de l'insuline / siRNA |
GenBank & GenPept Quelle différence y a-t-il entre les bases de données GenBank et GenPept ? |
GenBank Base de données des fichiers GenBank (extension ".gbk") de toutes les séquences de nucléotides. Fichier GenBank : voir le détail de son annotation/nomenclature (explication de chaque champs et sous-champs ou "Features"). |
GenPept Base de données des fichiers GenPept (extensions ".gp") de traduction (acides aminés) de toutes les séquences codantes ("CoDing Sequence" - CDS - nucléotides). Le format d'un fichier GenPept dérive de celui du fichier parent GenBank et son contenu est synchronisé avec chaque nouvelle version de ce fichier. |
2. Combien de fichiers GenPept le moteur de recherche "Entrez" renvoie-t-il avec le mot clé "protein" ? En combien de pages ces fichiers sont-ils répartis avec 200 résultats par page ? |
≈ 1,57 milliards de fichiers. ≈ 7,83 millons de pages. |
| 3. Effectuer les requêtes suivantes et conclure. | ||
| champs de la requête | mot-clé | résultats |
| protein | protein | ≈ 1,57 milliards |
| nucleotide | protein | ≈ 154 millions |
| protein | gene | ≈ 1,25 milliards |
| nucleotide | gene | ≈ 267 millions |
| A. Etude du pli Rossmann des déshydrogénase à NAD(P)+ |
1. Aller au NCBI. Rechercher les séquences protéiques de la lactate déshydrogénase. Attention : anglais, abréviation, emploi des opérateurs logiques (Booléens) : "AND", "OR" et "NOT". Avec l'option "Advanced" (lien en haut de la page), affiner la recherche avec EC 1.1.1.27 et Arabidopsis thaliana dans les résultats précédents : "#1 AND in builder" puis taper "1.1.1.27" avec le champs "EC/RN Number" du menu déroulant. Enregistrer la séquence au format FASTA du fichier AAC02678. |
≈ 894.500 résultats
1 séquence : AAC02678 |
Comment sait-on qu'il s'agit d'une enzyme ? Quelle réaction catalyse-t-elle ? Dans quelle voie métabolique ? |
|
Remarques
|
|
| En quoi le fichier de numéro d'accession AAN87112 est-il "abusif" ? En quoi n'est-il cependant pas "erroné" ? |
|
|
2. Ouvrir le fichier GenPept NP_002292. Examiner les informations des différents champs. Ouvrir l'onglet "Graphics" (en haut). Examiner la partie "site Features - CDD" Zoomer à 50% (loupes au centre) afin qu'apparaissent les lettres des acides aminés. Survoler les sites de fixation du NAD+ et du substrat avec les flèches larges (au centre). |
Quels acides aminés sont communs aux deux sites ? Pourquoi les acides aminés de chacun de ces sites ne sont-ils pas contigus dans la séquence ? |
Acides aminés du site de fixation du NAD+ :
Acides aminés du site de fixation du substrat :
Les acides aminés N138, H193 et T248 sont communs aux 2 sites de fixation : ils établissent donc des liaisons avec la molécule de coenzyme et avec la molécule de substrat. Chacun des sites de fixation résultent du regroupement d'acides aminés dans l'espace via le repliement de la chaîne polypeptidique et la formation de domaines indépendants. |
| 3. Cliquer sur le lien ci-contre : "Conserved Protein Domain Family - LDH1". | |
|
|
| Examiner l'alignement automatique généré en bas de la page "Conserved Protein Domain Family - LDH_1". Repérer un motif consensus GXGXXG dans la partie N-terminale des séquences. Ecrire l'expression régulière de ce motif dans la syntaxe Prosite. |
|
| Quelles sont les particularités physico-chimiques de la glycine ? |
|
Rechercher les statistiques de la base de données Expasy [UniProtKB/Swiss-Prot]. Quelle est la fréquence de cet acide aminé ? |
Données statistiques : G = 7,07% |
| Qu'en conclure ? |
|
Aller à InterPro. Rechercher "lactate dehydrogenase nad+ Arabidopsis". Pourquoi obtient-on autant de résultats ? |
Il y des résultats pour la lactate déshydrogénase et pour d'autres déshydrogénase. Certains résultats ont trait à des informations plus générales. Exemple : IPR036291 |
A quoi correspondent les informations de la colonne "Source database" ? Qu'est le consortium InterPro ? |
A certaines des bases de données du consortium InterPro. |
Dans la liste de résultats, afficher plus de 20 résultats par page. Quelle est l'expression régulière ? L'interpréter. |
Lien "View PS00064 in PROSITE patterns". Expression régulière : [LIVMA]-G-[EQ]-H-G-[DN]-[ST] |
| 4. Rechercher dans la base de données Uniprot le fichier correspondant au fichier GenPept obtenu au NCBI. |
|
Numéro d'accession : O49191 (O49191_ARATH) Un trés grand nombre d'informations supplémentaires. Notamment :
|
A quelle protéine correspond le fichier GenPept ABI54333 ? Vérifier avec le programme d'alignement de séquences MULTALIN si les séquences des fichiers AAC02678, O49191 et ABI54333 sont identiques (dans ce cas les valeurs du paramètre "check" au dessus de l'alignement obtenu sont identiques). Pourquoi le fichier ABI54333 n'est-il pas renvoyé dans la requête d'origine au NCBI ? |
ABI54333 : lactate déshydrogénase de Arabidopsis thaliana.
Le champs "DEFINITION" du fichier GenPept ABI54333 contient "At4g17260 [Arabidopsis thaliana]" et non "lactate dehydrogenase". |
Pourquoi le fichier ABI54333 n'est-il pas renvoyé dans la requête d'origine au NCBI ? |
Le champs "DEFINITION" du fichier GenPept ABI54333 contient les termes "At4g17260 [Arabidopsis thaliana]" et non les termes "lactate dehydrogenase". |
5. Aller à ScanProsite.
|
Lien vers le motif : PS00064 |
Quelle est l'expression régulière de ce motif ? Pourquoi est-elle identique à la précédente (voir ci-dessus) ? |
[LIVMA]-G-[EQ]-H-G-[DN]-[ST] Prosite fait partie du consortium InterPro |
6. Aller à PHI-BLAST au NCBI.
Sélectionnez (cocher) 10 résultats caractérisés par des E-value très différentes les unes des autres. Voir un descriptif de BLAST. |
7. Illustration : structure du domaine liant le NAD(P)+ - le pli Rossmann
Voir les spécificités du pli Rossmann des déshydrogénases à NAD(P)+ pour la suite. |
|
a. Visualisation de la lactate déshydrogénase de Squalus acanthias à une résolution de 3 Å Code PDB : 3LDH Le pli Rossmann ("Rossmann fold" - en hommage à Michael Rossmann) est une structure super-secondaire (assemblage de plusieurs types de structures secondaires) composée de 3 feuillets β liés à 2 hélices α de manière alternée (motif β-α-β-α-β). Un pli Rossmann peut fixer 1 nucléotide. Donc le domaine de fixation d'un dinucléotides (tel que NAD+ ou NADP+) contient 2 plis Rossmann appariés, chacun d'eux fixant l'un des nucléotides du co-facteur. |
8. Recherche de structures de déshydrogénases homologues avec un algorithme d'aprentissage automatique |
L'algorithme pLM-BLAST |
pLM-BLAST (Kaminski et al., 2023) est un modèle de langage protéique ("protein Language Model") ou pLM. |
Différence majeure entre pLM-BLAST et les différentes versions de BLAST ? |
pLM-BLAST repose sur une approche non supervisée ne nécessitant :
De plus, pLM-BLAST calcule des alignements globaux et locaux, ce qui permet d'identifier des domaines et des sous-domaines protéiques. pLM-BLAST étend ainsi le concept de BLAST en remplaçant les matrices de substitution invariantes (exemples : PAM250, BLOSUM62, ...) par des [similarités par résidu d'acides aminés] entre les intégrations protéiques ("per-residue similarities between protein embeddings"). La similarité entre une paire de résidus d'acides aminés donnée dépend ainsi entièrement du contexte, c'est-à-dire de l'ensemble des résidus de la chaîne polypeptidique. |
Déroulement de l'algorithme |
a. L'intégration globale d'une séquence d'acides aminés est représentée par une matrice de taille [n x m] où n et m sont, respectivement :
Remarque : le script "embeddings.py" de pLM-BLAST utilise les intégrations générées par des pLM de type T5 (exemples : prott5, famille ESM ou tout modèle utilisant la classe "AutoModel" de Transformers). b. pLM-BLAST calcule ensuite la matrice de substitution pour 2 séquences à comparer par la multiplication des matrices d'intégrations de ces 2 séquences. |
Application de pLM-BLAST à la LDH Aller à pLM-BLAST.
|
Interpréter les résultats de l'onglet "Hits". |
ECOD_002396946_e6cepD2 : | 2003.1.1.40 | 6CEP D:1-160
|
Cliquer sur le lien du 1er résultat de la colonne "Accession". |
| Comparaison avec la classification ECOD |
Qu'est ECOD ? |
ECOD ("Evolutionary Classification of protein Domains" - Cheng et al., 2014) propose plusieurs classifications des protéines de la PDB par homologie des structures de leurs domaines. Les domaines protéiques de ECOD sont regroupés en 5 niveaux : (A) Architecture; (X) Homologie possible; (H) Homologie; (T) Topologie; (F) Famille. |
Aller à ECOD browser.
Comparer avec les résultats obtenus avec pLM-BLAST. |
9. Superposition de 2 structures de LDH |
Aller à la page du calcul du RMSD entre 2 structures de protéines.
|
|
Que traduit le RMSD ? Que conclure de la valeur calculée ? |
RMSD ("Root Mean Square Deviation") : superposition des carbones α des 2 structures comparées. Plus la valeur RMSD est faible, meilleure est la superposition des structures, plus ces structures sont similaires. |
Quelles parties des 2 chaînes polypeptidiques se superposent le moins bien ? Pourquoi ? |
Les boucles. Les boucles sont très flexibles et donc mobiles : elles sont parfois mal résolues dans les structures obtenues par différentes techniques. |
| B. Etude des sites de fixation du calcium de la calmoduline (CaM) |
| 1. Exemple de motif écrit avec la syntaxe PROSITE ("Pattern syntax") : <A-x-[ST](2)-x(0,1)-{V} |
Ce motif se lit : "Ala en position N-terminale puis n'importe quel acide aminé puis 2 fois (Ser ou Thr) puis aucun acide aminé ou n'importe quel acide aminé puis n'importe quel acide aminé sauf Val." |
| Ecrire le motif suivant avec la syntaxe PROSITE : "4 Ser puis (Asp ou Ser) puis n'importe quel acide aminé puis (Asp ou Glu) puis (Glu ou Gly ou Val) puis 1 à 7 fois n'importe quel acide aminé puis (Glu ou Gly) puis 1 à 2 fois n'importe quel acide aminé puis 4 fois (Arg ou Lys)." |
S(4)-[DS]-x-[DE]-[EGV]-x(1,7)-[EG]-x(1,2)-[KR](4) Exemple : SSSSSDDEEEEKRKR |
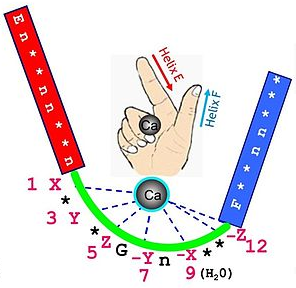
2. Le motif de fixation du calcium de la calmoduline s'appelle motif "EF-hand" composé d'environ 30 acides aminés :
La calmoduline fixe 4 atomes de calcium :
|
Source : PFAM (PF00036) |
Quelles sont les propriétés physico-chimiques des acides aminés de ce motif consensus ? Pourquoi ? |
Ces propriétés physico-chimiques permettent la fixation de Ca2+ (cation divalent). |
Ci-après, la séquence et la position des acides aminés du 1er motif "EF-hand" qui fixe le Ca2+de la calmoduline de Homo sapiens (exemple : CAA36839) : Retrouve-t-on le motif PROSITE ci-dessus ? |
La séquence FD21KD23GD25G ... KE32 répond à l'expression régulière du motif PROSITE. |
Visualisation de la calmoduline de l'homme (non complexée au calcium) à une résolution de 1,7 Å Code PDB : 1CLL Coordination et acides aminés du motif "EF - hand" EF1 : X = D21; Y = D23; Z = D25; -X = H2O; -Y = T27; -Z1 = E3; -Z2 = E32 |
3. Aller à ScanProsite. Rechercher avec l'option adaptée le motif consensus de fixation du calcium (motif "EF-hand") en utilisant l'expression régulière [DN]-x-D-G-[DN]-G-[TYQ]-x(4)-E. |
Analyser les résultats (exemples : "polymerase processivity factor component A20", "Calcium-binding allergen Bet v3", "Calbindin", ...). Quelles informations tire-t-on ? |
|
Repérer le résultat P84074.
|
|
4. Consulter le fichier PROSITE du motif "EF-hand" : PS00018 Récupérer le motif complet (attention au retour à la ligne / le point final n'en fait pas partie). |
Motif complet : D-{W}-[DNS]-{ILVFYW}-[DENSTG]-[DNQGHRK]-{GP}-[LIVMC]-[DENQSTAGC]-x(2)-[DE]-[LIVMFYW] |
Effectuer une recherche ScanProsite avec ce motif (choisir l'option 2). Fenêtre "STEP2 - Select a PROTEIN sequence database" :
Limiter le nombre de résultats à 1000 (option "Maximum number of displayed matches"). Pourquoi le résultat est-il étonnant ? |
Le motif est très long : le nombre de séquences qui y répondent est très grand. En conséquence, des protéines ou enzymes qui ne fixent pas le calcium ont malgré tout une partie de leur séquence qui correspondent à ce motif PROSITE. |
| C. Etude du récepteur de l'insuline |
a. Aller au NCBI. Effectuer une recherche de récepteur de l'insuline de l'homme.
|
Exemple : (insulin receptor[Protein Name]) AND Homo sapiens[Organism] On obtient beaucoup de fichiers correspondant :
|
Avec les fonctionalités de "Advanced", récupérer le sous-ensemble de fichiers qui ne contiennent pas le mot "partial". Combien de fichier(s) obtient-on ? Le fichier AAA59452 est intéressant.
Réfléchir à la requête la plus exacte (si il y en a une) pour n'obtenir que des fichiers de séquences complètes du récepteur de l'insuline de l'homme. |
Il n'y a pas de requête évidente à moins de consulter un grand nombre fichiers GenPept pour noter les mots à ne pas utiliser dans le filtre pour éviter d'éliminer des fichiers intéressants. |
b. Récupérer la séquence FASTA du fichier AAA59452 et effectuer une recherche de séquences homologues avec BLAST. |
|
Une nouvelle page s'ouvre. Comparer cette figure à celle du cours sur le récepteur de l'insuline. |
Ce récepteur est constituée d'un grand nombre de domaines. Un domaine est une région de la chaîne polypeptidique :
|
Cliquer sur le signe "+" de la fenêtre "List of domain hits" : on obtient la séquence des différents domaines. |
Le domaine PTKc_InsR. Domaine catalytique à activité protéine tyrosine kinase. |
Dans la figure du haut de la page, cliquer sur l'un quelconque des triangles oranges de la ligne intitulée "ATP binding site".
|
"Catalytic domain of the Protein Tyrosine Kinase, Insulin Receptor" "PTKs catalyze the transfer of the gamma-phosphoryl group from ATP to tyrosine (tyr) residues in protein substrates" Oui, dans la partie N-terminale des séquences. |
D. Interférence ARN Voir le cours sur l'interférence ARN. L'introduction d'ADN double brin ("double-strand DNA" - dsRNA) de plus de 30 nucléotides dans des cellules de mammifères induit la réponse interféron (activation de la protéine kinase R ("interferon-induced, double-stranded RNA-activated protein kinase") et de la 2',5'-oligoadénylate synthétase). Cette réponse entraîne la dégradation non spécifique des ARN messagers et une diminution du taux de traduction. La conception de siRNA ("design" / "screening") nécessite que les ARN synthétisés contiennent moins de 30 nucléotides. Les siRNA avec un débordement en 3’ constitué du dinucléotide UU sont les plus puissants.
|
Les règles de conception d’un siRNA à partir d’une séquence d’ARM messager sont :
EMBOSS Seqret : logiciel de conversion de formats de fichiers. |
Trouver la séquence d’un siRNA dans la séquence de l’ARN messager (GenBank NM_003380) codant la vimentine de l’homme : Uniprot : Homo sapiens vimentin mRNA |
>NM_003380.3 Homo sapiens vimentin mRNA CCCCGCGCCAGAGACGCAGCCGCGCTCCCACCACCCACACCCACCGCGCCCTCGTTCGCC TCTTCTCCGGGAGCCAGTCCGCGCCACCGCCGCCGCCCAGGCCATCGCCACCCTCCGCAG CCATGTCCACCAGGTCCGTGTCCTCGTCCTCCTACCGCAGGATGTTCGGCGGCCCGGGCA CCGCGAGCCGGCCGAGCTCCAGCCGGAGCTACGTGACTACGTCCACCCGCACCTACAGCC TGGGCAGCGCGCTGCGCCCCAGCACCAGCCGCAGCCTCTACGCCTCGTCCCCGGGCGGCG TGTATGCCACGCGCTCCTCTGCCGTGCGCCTGCGGAGCAGCGTGCCCGGGGTGCGGCTCC TGCAGGACTCGGTGGACTTCTCGCTGGCCGACGCCATCAACACCGAGTTCAAGAACACCC GCACCAACGAGAAGGTGGAGCTGCAGGAGCTGAATGACCGCTTCGCCAACTACATCGACA AGGTGCGCTTCCTGGAGCAGCAGAATAAGATCCTGCTGGCCGAGCTCGAGCAGCTCAAGG GCCAAGGCAAGTCGCGCCTGGGGGACCTCTACGAGGAGGAGATGCGGGAGCTGCGCCGGC AGGTGGACCAGCTAACCAACGACAAAGCCCGCGTCGAGGTGGAGCGCGACAACCTGGCCG |
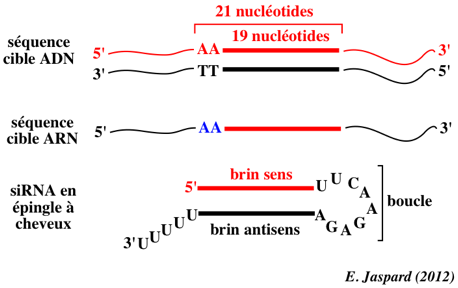
Il faut trouver une séquence de 21 nucléotides dans l’ARNm cible qui commence par un dinucléotide AA. On cherche le codon d'initiation de la transcription (AUG) : toutes séquences commençant par AA (et les 19 nucléotides suivants) constituent un site cible potentiel pour les siRNA. |
|
| Résultat pour la vimentine | |
| séquence ciblée - ADNc ("targeted region") | 5' AACTACATCGACAAGGTGCGCTT |
| siRNA sens | 5' CUACAUCGACAAGGUGCGCdTdT |
| siRNA antisens | 5' GCGCACCUUGUCGAUGUAGdTdT |
| Autres exemples | ||
| Lamine A/C SC : 5'AACTGGACTTCCAGAAGAACATC sens : 5'CUGGACUUCCAGAAGAACAdTdT antisens : 5'UGUUCUUCUGGAAGUCCAGdTdT |
Lamine B1 SC : 5'AACGCGCTTGGTAGAGGTGGATT sens : 5'CGCGCUUGGUAGAGGUGGAdTdT antisens : 5'UCCACCUCUACCAAGCGCGdTdT |
GL2 Luciferase SC : 5'AACGTACGCGGAATACTTCGATT sens : 5'CGUACGCGGAAUACUUCGAdTdT antisens : 5'UCGAAGUAUUCCGCGUACGdTdT |
| Source : Elbashir et al. (2001) | ||
Exemples de programmes IDT - "Custom Dicer-Substrate siRNA (DsiRNA)"
|