| Génomique fonctionnelle et protéomique : introduction |
| Tweet |
|
|
1. La génomique : présentation générale 2. Rappels et définitions 3. Les principaux buts de la génomique 4. Généralités sur le génome des plantes |
5. La protéomique 6. L'annotation : le prochain défi de la génomique 7. Les domaines en "omiques" 8. Liens Internet et références bibliographiques |
|
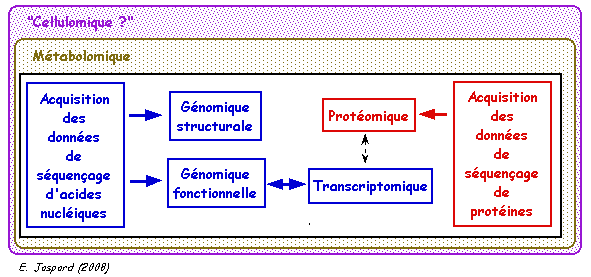
1. La génomique : présentation générale Même si les frontières sont floues, on peut proposer le découpage schématique suivant :
La génomique structurale, la génomique fonctionnelle, la transcriptomique et la protéomique sont des approches complémentaires.
Le "matériau" de base de la génomique est l'ensemble des séquences d'acides nucléiques et de séquences polypeptidiques obtenues par différentes méthodes de séquençage. Ces séquences et d'autres types d'informations qui découlent de leur analyse sont stockées dans des bases de données. L'accès aux bases de données s'effectue via le Web et Internet. Enfin, l'analyse de l'ensemble de ces données nécessitent des méthodes bioinformatiques. |
L'analyse des séquences d'ADN et de la structure des génomes ne permet pas d'associer directement une fonction à un gène. En d'autres termes, on ne peut pas inférer ce gène à :
Par ailleurs, tous les gènes ne s'expriment pas simultanément, ni au même taux.
En conséquence, afin de déterminer la fonction des ARN et des protéines associés à ces gènes, la génomique fonctionnelle analyse aussi :
|
|
Evolution des champs d'application de la génomique La génomique était à l'origine (fin des années 80) l'étude de la structure, du contenu et de l'évolution des génomes, en s'appuyant sur les résultats du séquençage de séquences nucléotidiques. Le domaine initial d'application de la génomique s'est élargi du fait :
Schématiquement, la génomique fonctionnelle a pour principaux buts de déterminer (liste non-exhaustive) :
*Attention : on confond souvent l'expression d'un gène, c'est-à-dire sa transcription (dans le noyau chez les Eucaryotes) et l'expression du produit de ce gène, c'est-à-dire l'activité biologique de ce produit (qui n'est pas destiné systématiquement, loin s'en faut, à rester dans le noyau). Le produit d'un gène est un (ou plusieurs) ARN. Si cet ARN est un ARN messager, le produit final est une (ou des) protéines. Plus précisément, la génomique fonctionnelle permet :
Les nouvelles techniques de séquençage en masse (ou massivement parrallèles ou à très haut débit) ont encore élargi le champs d'investigation de la génomique fonctionnelle. On peut citer (liste non-exhaustive) :
|
2. Rappels et définitions (principales sources : Unige-Ch / P. Luchetta et al. - 2005) a. Principaux types de gènes Il existe une trés grande diversité de types de gènes selon leur structure, leur localisation et leur fonction. On peut citer principalement :
|
|
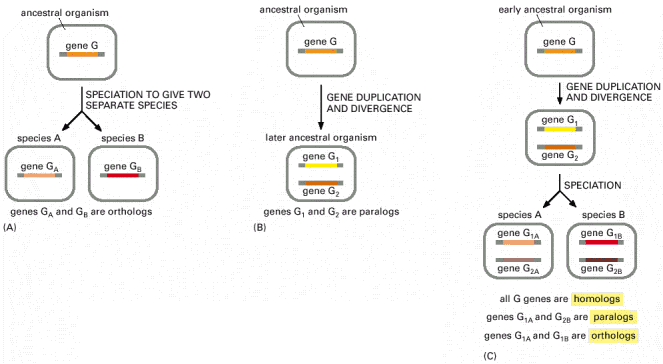
b. Gènes paralogues et gènes orthologues La duplication génique correspond à la multiplication de matériel génétique sur un chromosome. Des mutations peuvent alors affecter chaque copie des gènes après la duplication pour aboutir à une différenciation de deux gènes "frères" ou gènes paralogues. Donc, deux gènes au sein d'une espèce sont dits paralogues s'ils résultent d'une duplication génique. Lorsqu'une population évolue de manière indépendante (exemple : séparation géographique), ses caractéristiques génétiques évoluent de façon différente. Ce phénomène est appelé spéciation et il aboutit à la création de gènes orthologues : les mêmes gènes dans des espèces différentes. Donc deux gènes homologues (qui ont une forte similitude de séquence) de deux espèces différentes sont dits orthologues s'ils descendent d'un gène unique présent dans le dernier ancêtre commun aux deux espèces. Les espèces actuelles contiennent dans leur génome des gènes hérités d'un ancêtre commun (gènes homologues). Pour obtenir un arbre phylogénétique, on compare les gènes homologues. Quant un gène appartient à une famille multigénique, il est difficile de différencier les spéciations et les duplications. L'analyse des gènes paralogues est donc un élément important pour l'étude de l'évolution des génomes. Exemple de 2 types d'homologie de gènes basées sur des évènements évolutionnaires différents : (A) et (B) représentent les possibilités les plus simples. (C) représente un cas de figure plus complexe.
Source : "Molecular biology of the cell" |
|
c. Pseudogènes Ce sont des gènes non fonctionnels qui ont des similitudes avec un ou plusieurs gènes fonctionnels paralogues (gènes dupliqués au sein d'une même espèce). Ce sont donc des copies inactives de gènes fonctionnels. Cette inactivité est souvent due à l'absence de promoteur ou d'éléments de régulation. Les pseudogènes sont libres de toute contrainte sélective d'où une accumulation de mutations diverses dans leurs parties codantes ou non codantes. Il existe différents types de pseudogènes, notamment les pseudogènes dupliqués et les rétro-pseudogènes qui diffèrent par leur mécanisme d'apparition et leur mode d'évolution.
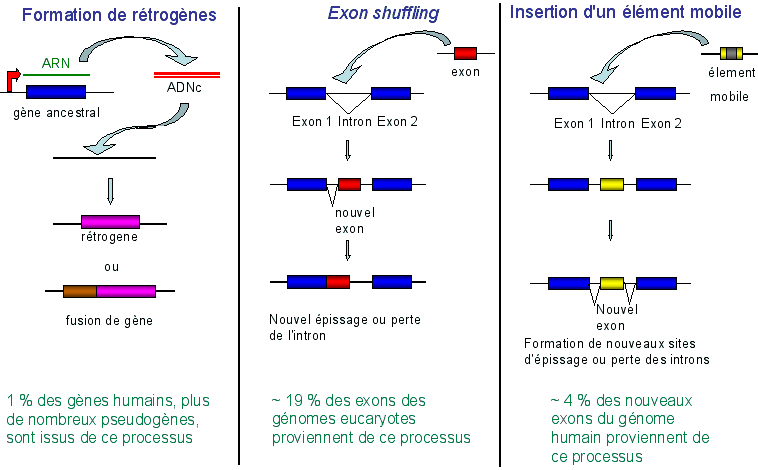
Les pseudogènes sont des indices d'évolution d'un organisme au cours du temps. Plus un organisme a évolué, plus grand est son nombre de pseudogènes. d. Rétroséquence Séquences qui résultent d'une transcription inverse puis qui sont intégrées au génome mais qui n'ont plus la capacité de se transposer. Elles ont certaines caractéristiques :
Les rétroséquences sont soit fonctionnelles (rétrogènes), soit non- fonctionnelles (rétro-pseudogènes). e. Rétrogène Le maintien de la fonction de ces séquences est un cas assez rare pour les raisons suivantes :
En conséquence, la majeure partie des rétroséquences sont des rétro-pseudogènes.
Source : B. Dujon (2008) |
| f. Eléments
transposables
Ceux sont des séquences d'ADN capable de se déplacer et de se multiplier de manière autonome dans un génome, par un mécanisme appelé transposition. Les transposons ont été identifiés par Barbara McClintock qui a étudié des mutations instables induites par ces éléments chez le maïs. Présents chez tous les organismes vivants, les éléments transposables sont un des constituants les plus importants des génomes eucaryotes. Exemples : 45% du génome de l'homme et plus de 70% chez le maïs. Les éléments transposables peuvent expliquer d'importantes différences dans les tailles du génome d'organismes. Les éléments transposables sont divisés en 2 classes selon leur mode de transposition. Ceux de classe I ou rétrotransposons : ils transposent via un intermédiaire ARN selon un mode réplicatif (copier - coller).
Leur ARN messager est rétro-transcrit en ADNc qui est intégré dans le génome. Les rétrotransposons sont divisés en 2 groupes :
Ceux de classe II ou transposons : ils transposent selon un mode conservatif (couper - coller).
Source : "La génomique végétale et les plantes cultivées" - M. Caboche Ils sont caractérisés par la présence à leurs extrémités de séquences terminales inversement répétées (TIR). Ils sont de taille variable (100 à 20 000 pb). Voir la base de données "ACLAME" : "A CLAssification of genetic Mobile Elements" |
| Eléments transposables | Arabidopsis thaliana | Homo sapiens | Saccharomyces cerevisiae |
| nombre de copies classe I - LTR | 1594 | 443.000 | 331 |
| nombre de copies classe I - non LTR | 515 | 2.426.000 | 0 |
| nombre de copies classe II | 2203 | 294.000 | 0 |
| g. Famille Alu
Famille de séquences répétées issue d'un processus de rétrotransposition. Les séquences Alu contiennent un site de reconnaissance pour l'enzyme de restriction Alu I.
La famille Alu est un exemple de SINE ("Short Interspersed Nuclear Elements" - voir ci-dessus). La plupart sont des rétroséquences issues d'une réverse transcription de l'ARN. h. La synténie Localement l'ordre des gènes sur un même chromosome tend à être conservé sur des millions d'années: c'est le phénomène de synténie. La synténie est donc la présence simultanée sur le même chromosome de deux ou plusieurs loci. La synténie est utilisée en génomique comparative (cartes physiques comparatives) pour décrire la conservation de l'ordre des gènes entre deux espèces apparentées. Les comparaisons entre espèces éloignées phylogénétiquement éloignées révèlent une perte de synténie. Cinteny - "Server for Synteny Identification and Analysis of Genome Rearrangement" i. Gène candidat C'est un gène dont on suppose l'implication dans un effet biologique. C'est donc un gène qui gouverne une part importante de la variabilité d'un caractère. Plusieurs approches (qui peuvent être combinées) existent pour identifier un gène candidat :
|
|
j. Les microsatellites Une séquence d'ADN dite microsatellite (ou "simple sequence repeats" - SSR, "short tandem repeats" - STR) est formée par une répétition continue de motifs de 2 à 10 nucléotides (exemple : le motif CAGT). Les microsatellites sont très abondants chez les Eucaryotes : un microsatellite peut être présent à des milliers d'exemplaires dans le génome. Chez les végétaux supérieurs, il y aurait en moyenne un microsatellite tous les 50 kb. Les microsatellites sont présents sur l'ensemble du génome, le plus fréquemment au niveau des introns et des exons des gènes. La localisation des microsatellites sur le génome est relativement conservée entre des espèces phylogénétiquement proches. Le polymorphisme des microsatellites peut être utilisé comme marqueur génétique afin d'identifier un individu. Les régions flanquantes des microsatellites servent d'amorces pour la réaction de polymérisation en chaîne (PCR). En effet, si un microsatellite donnné n'est pas spécifique d'un locus, en revanche ses régions flanquantes le sont : une paire d'amorces spécifique de ces régions flanquantes permettra donc l'amplification spécifique de ce seul microsatellite. k. Marqueur génétique C'est une séquence d'ADN polymorphe utilisée pour baliser le génome et obtenir une carte génétique. Les nouvelles biotechnologies permettent l'analyse directe du polymorphisme des séquences d'ADN. Il existe différentes sortes de marqueurs :
|
|
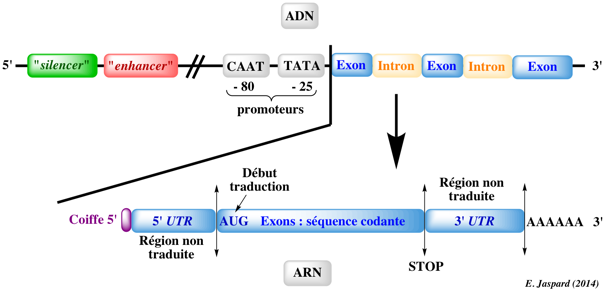
3. Les principaux buts de la génomique a. L'assemblage de cartes physiques et génétique des génomes : voir un exemple La position des gènes dans un génome peut être définie soit par une distance physique, soit par une position relative basée sur des fréquences de recombinaison entre ces gènes. Cette information est capitale si l'on veut comparer les génomes d'espèces voisines ou établir le lien entre données phénotypiques et génotypiques. Les cartes génétiques décrivent l'ordre relatif des marqueurs génétiques au sein d'un groupe de liaison. Les marqueurs peuvent être de différentes natures : gènes, microsatellites, SNP, EST, STS, RFLP ... Exemple : marqueur STS de la glutamate déshydrogenase. [dbSTS: database of "Sequence Tagged Sites"] Les cartes génétiques sont utilisées en recherche fondamentale mais aussi pour l'amélioration des espèces animales et végétales. b. L'obtention de séquences génomiques, de séquences transcrites et leur assemblage Initialement (dans les années 80 - 90), la méthode de séquençage développée par Frédéric Sanger a été utilisée puis trés largement automatisée tant pour pour les réactions de séquençage que pour la lecture des séquences. Puis l'avènement des technologies de séquençage à très haut débit a bouleversé la portée des résultats que l'on peut obtenir en génomique. Il y a 2 démarches pour le séquençage de génomes entiers : la méthode hiérarchique et la méthode dite "en vrac". Elles nécessitent l'assemblage des séquences chevauchantes en séquences sans interruption que l'on appelle contigs. Par ailleurs, la plus grande partie d'un génome Eucaryote étant constitué d'ADN non codant, le séquençage porte aussi sur des clones d'ADNc (pleine longueur ou non) qui contiennent des séquences issues de la transcription inverse d'ARN messagers. Le séquençage d'une des extrémités d'un ADNc suffit en principe pour identifier un clone d'ADNc et l'on appelle ces petits fragments de séquences des marqueurs de séquences exprimées ou EST ("Expressed Sequence Tags"). Le séquençage recquiert des logiciels bioinformatiques adaptés à la quantité phénoménale d'information qu'il génère. |
c. La fabrication d'atlas d'expression génique En étudiant les profils de transcription et de traduction, on peut cerner la fonction d'un gène. Les méthodes actuelles de génomique sont basées sur la détection de marqueurs spécifique de chaque gène dans une banque contenant des centaines de milliers de fragments séquencés. Pour découvrir de nouveaux gènes, on se sert du séquençage d'EST ou de méthodes telles que l'analyse sérielle de l'expression des gènes ("Serial Analysis of Gene Expression", méthode SAGE) ou bien encore celle de l'expression différentielle d'ARNm ("differential display"). Quand on dispose d'une collection d'EST spécifiques d'un gène ("unigene set") on peut fabriquer des puces à ADN ("micro-array", "chips") sur lesquelles sont hybridés des ADNc marqués par des molécules fluorescentes. Ces ADNc sont extraits à partir de tissus, de cellules auxquels on a appliqué un traitement particulier. La comparaison de l'expression des gènes par rapport à un témoin non traité permet théoriquement d'évaluer le niveau relatif de transcription de l'ensemble des gènes d'un génome dans des centaines de conditions. On peut également obtenir des informations sur la régulation de leur transcription, voire des indices sur la fonction de gènes inconnus, par comparaison avec des gènes de fonction connue. d. La collecte de données fonctionnelles sur les aspects biochimiques et sur l'action phénotypique des gènes La génomique fonctionnelle inclut des approches qui permettent de vérifier les propriétés biochimiques et les rôles cellulaires du produit de chaque gène. La génétique inverse (qui va du gène vers le phénotype) à haute densité consiste à inactiver de manière ciblée et systématique des gènes particuliers. Parmi les stratégies employées, on peut citer
e. L'évaluation de la variabilité de la séquence d'ADN au sein d'une même espèce (SNP - NCBI) Les génomes sont polymorphes, c'est-à-dire que 2 ou plusieurs variants de séquences différentes peuvent co-exister au sein d'une population naturelle. Le polymorphisme d'un seul nucléotide ou SNP ("Single nucléotide polymorphism") ou le polymorphisme d'insertion ou de délétion de nucléotides ont une part essentielle dans la variabilité génétique (la composante héréditaire de variabilité de caractères). Connaître la répartition des SNP est donc capital pour établir les associations entre ces variations SNP et les variations phénotypiques. |
|
f. L'identification et l'annotation de l'ensemble des gènes des génomes et de leurs produits d'expression : voir un exemple Après avoir séquencé un génome, il faut identifier les gènes qu'il contient. Plus précisément, cela consiste à :
Une fois un gène identifié, il faut l'annoter, c'est- à-dire le relier aux maximum de données biologiques (par exemple : données de génétique concernant sa fonction, son expression et les variations phénotypiques des mutants pour la protéine codée, ...). En d'autres termes, on tente d'assigner aux molécules pour lesquelles les gènes codent :
Des logiciels bioinformatiques sont nécessaires pour l'étude de la structure des gènes, Par exemple :
Des bases de données regroupent l'ensemble des données biologiques informatives quant à la structure et la fonction des gènes et elles permettent leur annotation :
Bien sur, l'ensemble de ces données sont intégrées dans les grandes bases de données biologiques mondiales que sont :
g. Le développement d'outils bioinformatiques. L'étude des génomes, des transcriptomes et des protéomes, des interactomes, nécessite le développement de technologies informatiques (Internet, ordinateurs, ...), de logiciels ou d'ensemble de logiciels et de théories informatiques, afin :
Toutes ces approches contribuent à élucider les mécanismes moléculaires qui régissent les fonctions biologiques dans une cellule. Elles restent cependant complémentaires des approches expérimentales "classiques" de la biochimie, de la biologie cellulaire, de la génétique et d'autres disciplines. |
|
4. Généralités sur le génome des plantes Les génomes des plantes contiennent plusieurs classes de gènes qui sont absents ou sous-représentés chez les animaux. Par exemple, les gènes codant pour :
Les classes de gènes communs aux plantes et aux animaux sont celles impliquées dans les mécanismes les plus généraux de la biologie d'une cellule : Il y a cependant des exceptions : par exemple, il n'y a pas d'homologue de la famille de protéines G Ras chez Arabidopsis thaliana. Les projets de séquençage de génomes de plantes portent sur un trés grand nombre d'espèces. a. Le génome d'Arabidopsis thaliana est l'un des modèles pour le règne végétal. Il sert de référence pour mettre au point des méthodes d'analyse fonctionnelle. Le génome d'Arabidopsis thaliana a été trés largement façonné par des duplications à grande échelle suivies par de nombreuses délétions et duplications en tandem. La situation actuelle des familles de gènes telle qu'on peut l'observer dans les séquences assemblées des cinq chromosomes d'Arabidopsis thaliana rend compte des pressions évolutives qui se sont exercées sur la fonction de chaque gène. b. Medicago truncatula, proche de la luzerne cultivée, a été choisi comme légumineuse modèle. Elle est diploïde et autogame, avec un petit génome. C'est un modèle utilisé pour la génétique moléculaire de la symbiose fixatrice d'azote avec la bactérie du sol Rhizobium et la symbiose endomycorhizienne. c. Du point de vue économique, les plus importants concernent les principales céréales : maïs, riz, blé, sorgho, orge et les plantes fouragères comme le soja et la luzerne. Les projets sur le maïs, le riz et la luzerne concernent les ressources génétiques quantitatives. Les caractères génétiques importants sur le plan économique sont, entre autre :
Un grand nombre de consortium regroupent l'ensemble des données (soit pour plusieurs espèces, soit par espèce) dans des banques de données. Par exemple :
|
|
La protéomique a pour but d'identifier et quantifier l'ensemble des protéines synthétisées ou protéome, à un moment donné et dans des conditions données au sein d'un tissu, d'une cellule ou d'un compartiment cellulaire. Le protéome est extrêmemement complexe à plusieurs titres :
La transcriptomique analyse l'ensemble des transcrits (produits d'expression des gènes). La protéomique et la transcriptomique sont donc des approches complémentaires trés puissantes qui peuvent être utilisées pour des études fondamentales ou appliquées en biologie, en médecine, en agriculture. En effet, dans les deux cas, les infomations recueillies permettent d'aborder l'ensemble des réponses cellulaires dans leur globalité et non plus de manière partielle. La protéomique apporte des réponses auxquelles la transcriptomique ne peut répondre :
|
6. L'annotation : le prochain défi de la génomique (et des domaines en "omique") L’annotation d’un génome, d'un transcriptome, d'un protéome, d'un métabolome ... consiste à documenter de la manière la plus exhaustive tous les composants de cette information brute. On conçoit que c'est un travail encyclopédique colossal, d'autant que de nouvelles données sont obtenues de plus en plus rapidement et massivement et que ces données peuvent être croisées. a. L’annotation automatique s'appuie (essentiellement) sur des comparaisons des séquences à annoter avec les séquences présentes dans les banques de données. Les algorithmes recherchent des similarités / homologies de séquence, de structure, de motifs, … Ils permettent de prédire la fonction d’une molécule et de transfèrer automatiquement l'annotation entre les molécules homologues. Mais il y a un point capital : si l'annotation des molécules de référence est correcte, il n'y a pas de souci. Si elle est fausse, c'est un "jeu de domino" : l'erreur initiale est répercutée de proche en proche. b. L’annotation manuelle (ou curation) par des experts (des curateurs) qui valident ou invalident la prédiction en fonction de leurs connaissances ou de résultats expérimentaux. L'annotation manuelle est donc tout à fait indispensable. Mais, en regard de la quantité phénoménale de données acquises quotidiennement, il est illusoire d'envisager une curation manuelle de l'ensemble des données en temps réel. On mesure aisément le problème : une quantité minime de données traitées par l'homme en temps réel qui induit un retard / décalage de plus en plus grand. c. L'annotation structurale dans le cas d'un génome tente de prédire :
Il existe des méthodes intrinsèques ou ab-initio qui s'appuient sur des techniques informatiques d'apprentissage automatique utilisant :
Il existe des méthodes extrinsèques qui reposent sur la comparaison des cadres de lecture avec les séquences présentes dans les banques de données (exemples : "Orpheus", "Critica", "Reganor", ...). d. L'annotation fonctionnelle tente de prédire la fonction potentielle des gènes (notion d'étiquette, avec nom, fonction et interactions probables). e. L'annotation relationnelle tente de décrire les relations (interactions) entre les produits des gènes (familles de gènes, réseaux de régulation, réseaux métaboliques, ...). Toutes ces démarches d'annotation vont de paire avec : (a) Le développement d'une ontologie : un recueil de termes soigneusement sélectionnés afin de standardiser la dénomination de 3 concepts fondamentaux. Ainsi, l'ontologie du consortium "Gene ontology" (GO) contient environ 38 000 termes ("Cellular Component" : 3200 termes, "Molecular Function" : 9600 termes, "Biological Process" : 25000 termes). Ces termes sont placés dans une hiérarchie rigoureuse qui établit des liens univoques entre eux. (b) L'utilisation de langages spécifiques pour standardiser le format des données afin qu'ils soient transmis sans difficulté d'un service (logiciel) bioinformatique à un autre. De plus en plus, le langage XML avec des schémas de format XSD s'imposent. La transformation d'un type de données en un autre est d'autant plus performante que les formats en entrée et en sortie sont standardisés afin que n'importe quel logiciel puissent "accepter" les données entrantes (format d'échange commun BioXSD). |
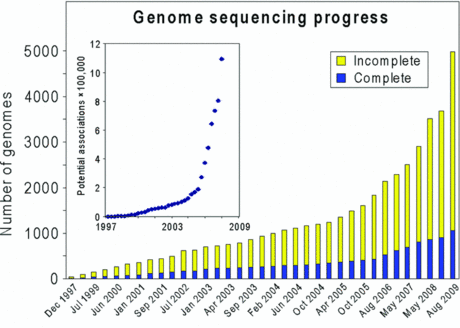
Décalage entre le nombre de génomes séquencés et leur documentation encyclopédique On peut considérer que l'annotation est un point d'achoppement des domaines en "omique". En regard de la performance extraordinaire des technologies de ces domaines (on envisage trés prochainement le séquençage d'un génome par jour) et donc l'accumulation tout à fait colossale de données de tous types dans les banques de données, on ne peut qu'induire un décalage entre les données brutes et leur interprétation, donc leurs significations biologiques.
Source : Hanson et al. (2010) Voir un exemple de "méta-données" ("data") selon l'ontologie de EDAM ("EMBRACE Data and Methods"). Tant que ce décalage existera, le pas suivant qui est l'extraction d'une "sur-information biologique" de cette information brute, ne pourra être complètement franchi. |
| Evolution du nombre de génes estimés dans le génome humain | |||
| Technique | Date | Nombre de gènes estimés | Hypothèses et commentaires |
| "Calcul" initial | 1990 | 100 000 | Avec l'hypothèse que la taille moyenne d'un gène = 30 kb |
| Ebauche de séquençage du génome | 1994 | 71 000 | Résultat biaisé par les régions riches en gènes ? |
| Ilôts CpG | 80 000 | Avec l'hypothèse que 66% des gènes humains ont de tels "ilôts" | |
| Analyse des EST | 1994 | 64 000 | Gènes ayant un homologue dans GenBank - Redondance des EST de 50% |
| Chromosome 22 | 1999 | 45 000 | Correction liée à la haute densité en gène de ce chromosome |
| Technique "Exofish" ("Exon Finding by Sequences Homology") | 2000 | 28 000 - 34 000 | Avec l'hypothèse que les régions codantes sont plus conservées que les non-codantes. Comparaison des génomes homme - poisson ("Tetraodon nigroviridis") |
| EST | 2000 | 35 000 120 000 |
Nombre de gènes Nombre de transcrits |
| Premier "brouillon" du génome | 2001 | 30 000 - 40 000 | Gènes connus + prédictions |
| Comparaison avec le génome de la souris | 2002 | 30 000 | Gènes connus + prédictions |
| Génome abouti | 2004 | 20 000 - 25 000 | Gènes connus + prédictions |
| Génome abouti | 2007 | 20 000 | Annotation des gènes améliorée |
Consortium ENCODE |
2012 | Publication de 30 articles qui montrent entre autre :
|
|
| Source : Duret L. (2011) - "Bioinformatique: Annotation des génomes (eucaryotes)" | |||
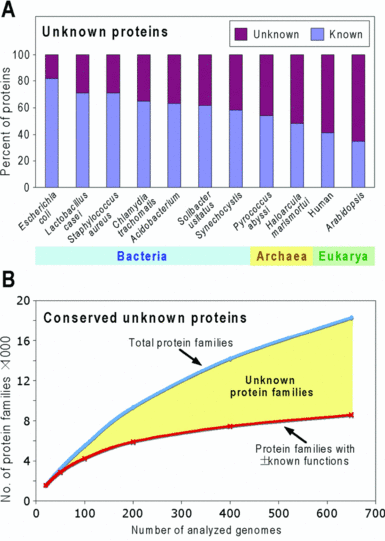
Les erreurs ou l'absence d'annotation Les "inconnues" dans les bases de données :
Source : Hanson et al. (2010)
Il est dommage d'accumuler une quantité inouie de données si on ne peut pas en tirer toute l'information. Ce déluge de données peut même noyer l'information actuelle pertinente et nuire (au moins dans un premier temps). Quel intérêt d'obtenir à la suite d'une étude longue et coûteuse via des EST ou des puces à ADN, des informations telles que : "not annotated", "hypothetical protein", "unnamed molecule", "putative function". On aboutit aux même conclusions : X gènes sont sur-exprimés et Y sont sous-exprimés. Mais qui sont-ils, que font-ils, où sont adressées les protéines pour lesquelles ils codent ... ? Ci-dessous, protocole d'analyse des erreurs d'annotation des fonctions des super familles d'enzymes dans les bases de données publiques. |
|
Les autres moyens pour l'annotation L'ensemble de ces moyens fait partie de la génomique comparative et s'appuie sur la notion d'association. L'exemple typique est celui de génes bactériens regroupés en opéron codant les différentes étapes d'une voie métabolique : la fonction d'un géne inconnu peut-être inferrée à partie des génes connus de cet opéron. Cette notion est étendue à la comparaison de génomes entiers. Une étude récente a ainsi permis de prédire la fonction de 19 familles de protéines d'Arabidopsis et de Procaryotes (Gerdes et al., 2011). Parmi ces moyens, on peut citer :
|
| Exemples de systèmes d'annotation de génomes, de gènes, de voies métaboliques, de réseaux d'interactions etc ... | |
| "The gene ontology (GO) database" | GO current annotations |
| KAAS ("KEGG Automatic Annotation Server") | Moriya et al. (2007) "KAAS: an automatic genome annotation and pathway reconstruction server" Nucleic Acids Res. 35, W182-W185 |
| "The Joint Genome Institute's (JGI) Integrated Microbial Genomes (IMG) system" | Markowitz et al. (2009) "The integrated microbial genomes system: an expanding comparative analysis resource" Nucleic Acids Res., 1-9 |
| "The National Microbial Pathogen Data Resource's (NMPDR) Rapid Annotation using Subsystems Technology (RAST) server" | Aziz et al. (2008) "The RAST Server: Rapid Annotations using Subsystems Technology" BMC Genomics 9, 75 |
| "J. Craig Venter Institute (JCVI) Annotation Service" "The Glimmer system" : suite logicielle pour le séquençage et l'assemblage de génomes, la recherche de gènes, l'annotation et l'analyse de génomes, l'analyse métagénomique (et autres outils génomiques et protéomiques) |
"The Glimmer system" |
| RGAP : "Rice Genome Annotation Project" | |
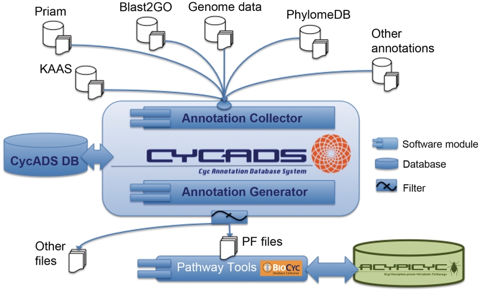
| "CycADS : an annotation database system to ease the development and update of BioCyc databases" BioCyc : ensemble de bases de données (près de 3000 génomes et leurs voies métaboliques) Source : Vellozo et al. (2011) |
 |
| 7. Liens Internet et références bibliographiques | |
| "Précis de Génomique" (2004) G. Gibson & S. Muse - Ed. De Boeck - ISBN : 2-8041-4334-1 | |
|
Numéro spécial sur les bases de données dédiées aux plantes : Plant Physiol. 135 (2005) "La génomique végétale et les plantes cultivées" - Michel Caboche - Conférence "Université de tous les savoirs" (Vidéo et documents à télécharger) Schoof et al. (2004) "MIPS Arabidopsis thaliana Database (MAtDB): an integrated biological knowledge resource for plant genomics" Nucleic Acids Res. 32 - Database issue: D373 - 376 "AraCyc" : Arabidopsis thaliana Biochemical Pathways "The Plant Specific Database" - Université du Michigan |
|
|
Bakke et al. (2009) "Evaluation of Three Automated Genome Annotations for Halorhabdus utahensis" PLoS ONE 4, e6291 Schnoes et al. (2009) "Annotation error in public databases: misannotation of molecular function in enzyme superfamilies" PLoS Comput. Biol. 5, e1000605 Hanson et al. (2010) "‘Unknown’ proteins and ‘orphan’ enzymes: the missing half of the engineering parts list – and how to find it" Biochem. J 425, 1-11 Gerdes et al. (2011) "Synergistic use of plant-prokaryote comparative genomics for functional annotations" BMC Genomics 12, S2 Kubrycht et al. (2012) "Virtual Interactomics of Proteins from Biochemical Standpoint" Mol. Biol. Int. |
|
|
"BioCyc" : ensemble de bases de données. Près de 3000 génomes et leurs voies métaboliques. "FunCoup" : base de données d'interactions protéiques Vellozo et al. (2011) "CycADS: an annotation database system to ease the development and update of BioCyc databases" Database |
|
![]()