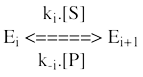| Les réactions enzymatiques à 2 (ou plus) substrats |
| Tweet |
|
|
1. Introduction et définitions 2a. Système séquentiel (ou à simple déplacement) appelé au hasard 2b. Démarche expérimentale pour la détermination des vitesses initiales 3. Système séquentiel (ou à simple déplacement) appelé ordonné 4. Détail du mécanisme séquentiel ordonné de l'acyltransférase LpxD 5. Système à double déplacement appelé Ping Pong 6. Mécanismes d'inhibition des systèmes à 2 substrats
|
7. Mécanismes plus complexes
8. Utilisation d'isotopes pour distinguer les mécanismes : échange isotopique et effets des isotopes 9. Cas des protéases à sérine 10. Liens Internet et références bibliographiques |
|
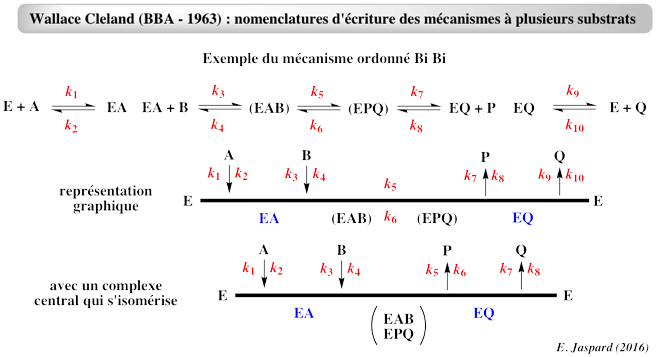
1. Introduction et définitions Nous faisons l'hypothèse de l'établissement rapide des différents équilibres de fixation entre l'enzyme et les substrats :
Pour ce second type de système, l'hypothèse de l'état stationnaire est préférable (voir "Modèles homéomorphes") : Cependant, cette approche nécessite une méthode graphique qui permet de représenter la distribution des différents complexes enzymatiques en termes de concentration de substrat et des différentes constantes de vitesse : la méthode de E. King & C. Altman (1956). De plus, l'équation de la vitesse ainsi obtenue n'est pas encore utilisable telle quelle tant que les constantes de vitesse n'ont pas été exprimées en termes de paramètres cinétiques déterminables expérimentalement (VM, KM et KI). Il faut alors appliquer les règles générales (ou nomenclatures) développées par Wallace Cleland (1963).
Les mécanismes à plusieurs substrats sont de 3 types :
|
| Définitions | |
|
Substrats Produits Enzyme |
|
| Forme enzymatique |
|
|
Complexe central |
|
|
Complexe terminal (ou abortif) |
|
| Séquentiel |
|
| Non séquentiel |
|
| Theorell - Chance |
|
|
Nomenclature des systèmes |
|
| Modèles homéomorphes |
|
| Postulat 1 |
|
| Postulat 2 |
|
|
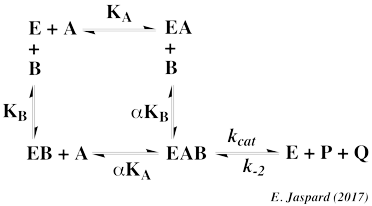
2. Système séquentiel (ou à simple déplacement) appelé au hasard Deux substrats, A et B, se fixent de manière aléatoire sur l'enzyme libre E (c'est-à-dire qu'il n'y a pas de fixation privilégiée de l'un ou l'autre des deux substrats) avec une constante de dissociation KA et KB, respectivement. Parfois la fixation de l'un des deux substrats modifie la constante d'équilibre de dissociation de l'autre substrat de l'enzyme libre d'un facteur α. Le mécanisme réactionnel s'écrit :
On aboutit à un ensemble de courbes de saturation pour chaque substrat (voir la démonstration des équations : vi = f([A0]) et vi = f([B0]).
|
|
Représentations graphiques obtenues avec le mécanisme au hasard Les paramètres : α, KA et KB sont déterminés à partir :
La description de la démarche expérimentale pour déterminer les vitesses initiales permet de mieux comprendre ces deux types de représentations graphiques.
Voir un document qui explique la procédure simple pour tracer les droites des doubles inverses. |
Exemples d'enzymes qui ont un mécanisme catalytique au hasard
|
|
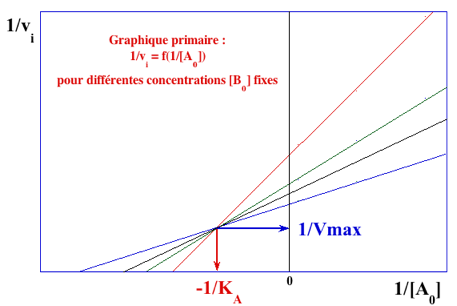
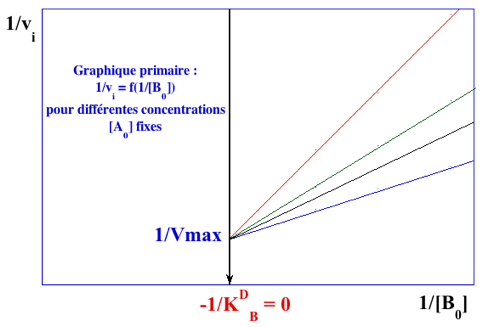
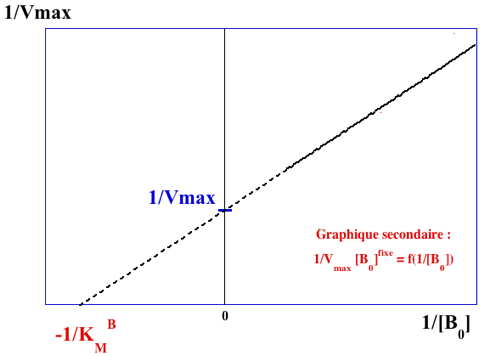
Représentations graphiques obtenues avec le mécanisme ordonné Le paramètre KA est confondu avec le paramètre KMA. Il est (sont) déterminé(s) à partir du graphe primaire pour le substrat A (ci-dessous) :
Les valeurs du graphe primaire pour le substrat A sont reportées dans le graphe secondaire pour le substrat B (ci-dessous) qui permet de déterminer : VM et KMB.
Voir un document qui explique la procédure simple pour tracer les droites des doubles inverses. |
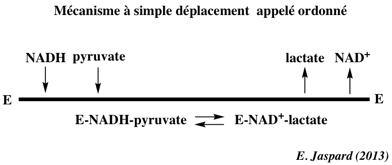
Exemples d'enzymes qui ont un mécanisme catalytique ordonné a. Les déshydrogénases à NAD+ : dans le cas de la lactate déshydrogénase (mécanisme Bi Bi ordonné), le coenzyme se fixe toujours en premier et le lactate est toujours relargué en premier.
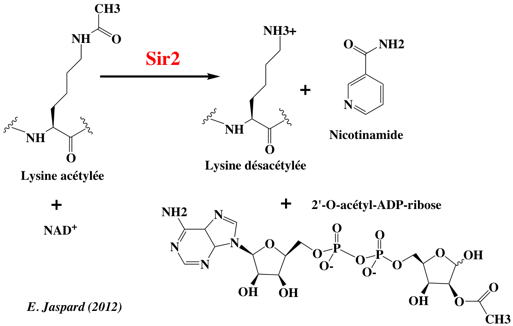
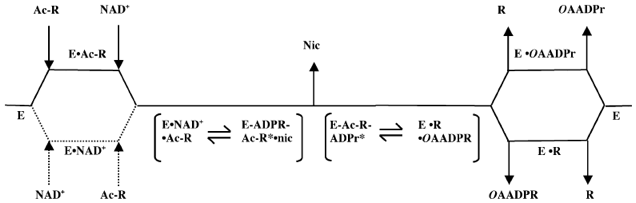
b. L'adénylate kinase (E.C. 2.7.4.3) catalyse la réaction globale : AMP + ATP <=> 2 ADP selon un mécanisme séquentiel ordonné. c. Les sirtuines sont des histones désacétylases nictotinamide adénine dinucléotide (NAD+)-dépendantes / ADP-ribosyltransférases. Elles couplent l'hydrolyse du NAD+ et la désacétylation d'un substrat acétylé pour former la nicotinamide, le produit désacétylé et le nouveau métabolite O-acétyl-ADP-ribose (OAADPR).
Figure ci-dessous : schéma réactionnel des sirtuines 2 qui suivent un mécanisme Bi Ter ordonné.
Source : Borra et al. (2009)
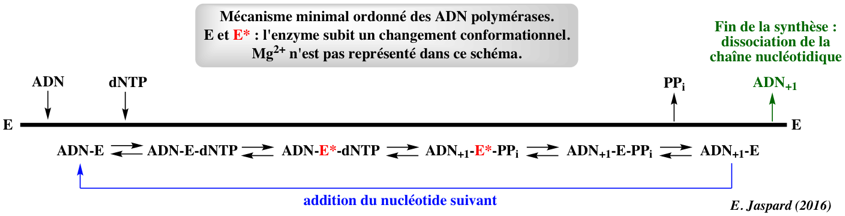
d. Mécanisme ordonné des ADN polymérases
|
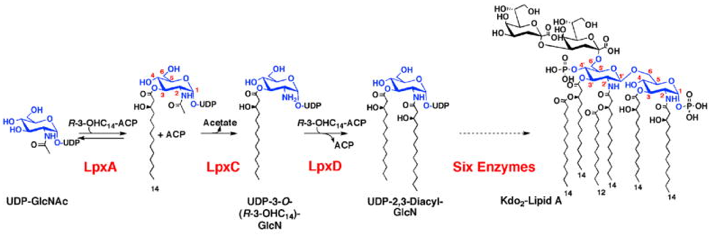
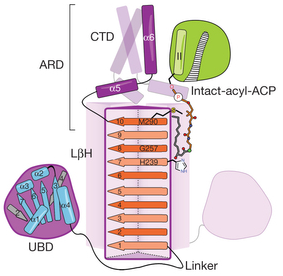
4. Détail du mécanisme séquentiel ordonné de l'acyltransférase LpxD Le lipide A (glycolipide phosphorylé) est la partie hydrophobe du lipopolysaccharide qui constitue le feuillet externe de la membrane externe de la plupart des bactéries gram négatives. Le lipide A ancre le lipopolysaccharide dans la membrane externe de la cellule et est généralement nécessaire pour la croissance bactérienne. La LpxD acyltransférase (UDP-3-O-(3-hydroxymyristoyl)glucosamine N-acyltransférase - EC 2.3.1.191) est la 3ème enzyme de la voie de biosynthèse du lipide A chez Escherichia coli. La LpxD catalyse la N-acylation [R-3-hydroxymyristoyl-ACP]-dépendante de l'UDP-3-O-(R-3-hydroxymyristoyl)-α-D-glucosamine pour former l'UDP-2, 3-diacylglucosamine et l'ACP. La chaîne acyle R-3-hydroxymyristoyl est portée par l'ACP.
Source : Bartling & Raetz (2008) Abréviations dans la figure ci-dessus:
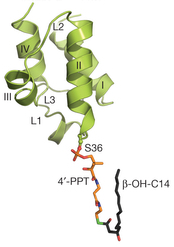
Figure ci-dessous : acyl-ACP intacte.
Source : Masoudi et al. (2013) L'ACP contient 4 hélices (I à IV) et des boucles (L1 à L3) 28, 29. Le bras 4′-PPT et la chaîne acyle R-3-hydroxymyristoyl sont indiqués. Chaque monomère du trimère de LpxD contient 3 domaines :
Source : Masoudi et al. (2013)
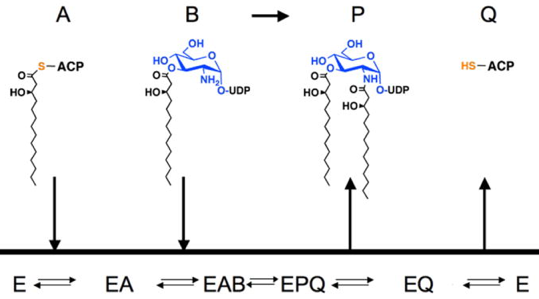
La zone de reconnaissance (interaction) de l'ACP (ARD : "ACP Recognition Domain") est constitué du domaine C-terminal et de la dernière hélice du domaine LβH. La LpxD suit un mécanisme Bi Bi ordonné dans lequel :
Source : Bartling & Raetz (2008)
|
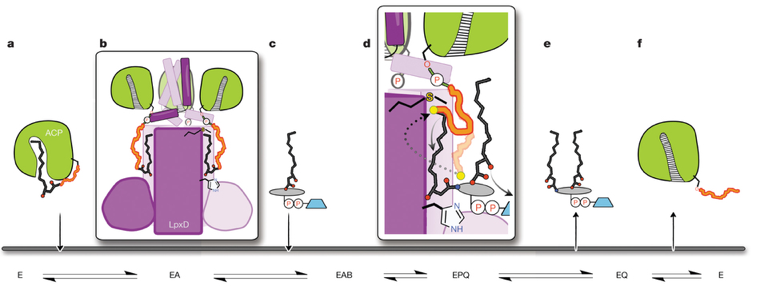
Explication structurale de ce mécanisme
Source : Masoudi et al. (2013) Figure a : l'acyl-ACP se fixe en premier à la forme libre de LpxD, formant le complexe binaire (EA - figure b). Figure b : l'ACP est associée à l'ARD ("ACP recognition domain") et le groupe acyle 4 '-phosphopantéthéine (4′-PPT), lié à Ser 36, est empaqueté dans un canal hydrophobe. Figure c : l'UDP-acyl-GlcN se fixe ensuite ce qui initie le transfert du groupe acyle. Figure d : au sein du complexe ternaire, le bras de 4′-PPT issu de l'hydrolyse de l'acyl-ACP (ligne ondulée orange transparente) englobe complètement la chambre catalytique, ce qui empêche l'UDP-diacyl-GlcN de se dissocier. Le déplacement de 4′-PPT (ligne ondulée orange foncée) vers Met290 (flèche en pointillés) ouvre la chambre catalytique. Figure e : ce mouvement entraîne la sortie éventuelle de l'UDP-diacyl-GlcN. Figure f : ce mouvement déclenche des changements conformationnels en aval de l'hélice II conduisant à une dissociation de l'holo-ACP. |
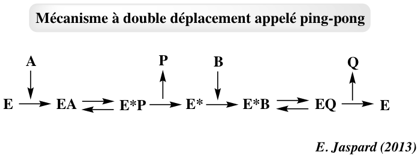
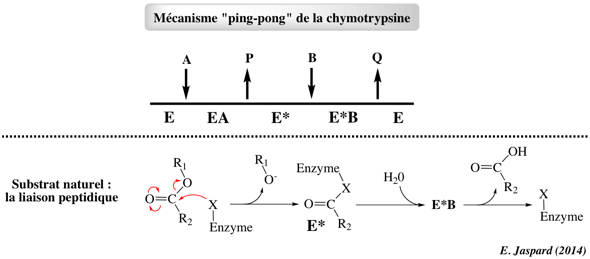
5. Système à double déplacement appelé "Ping Pong" Un ou plusieurs produits est/sont relargué(s) avant que tous les substrats ne soient fixés à l'enzyme. Ce mécanisme est caractérisé par la formation obligatoire d'une forme intermédiaire modifiée de l'enzyme (E*) qui est un acyl-enzyme.
Exemples d'enzymes
|
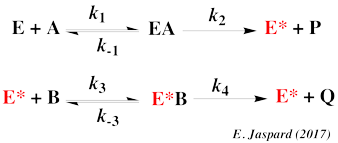
Modèle cinétique du mécanisme "Ping-Pong" Le mécanisme réactionnel s'écrit :
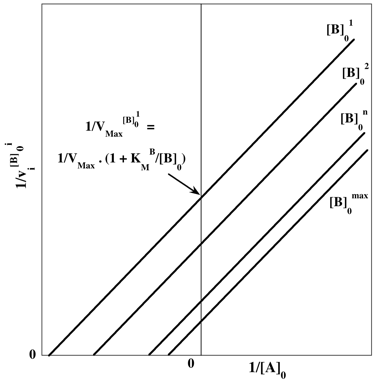
Ce mécanisme est caractérisé par un ensemble de droites parallèles selon la représentation en double - inverses :
|
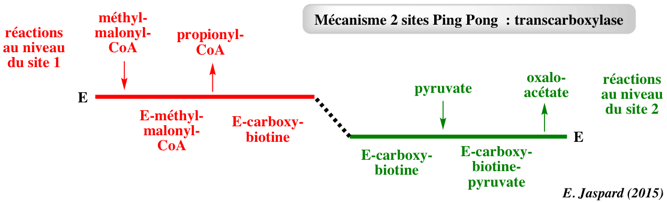
Mécanisme "Ping Pong" à 2 sites (ou plus) La transcarboxylase (EC 2.1.3.1) catalyse un mécanisme appelé Ping Pong à 2 sites ("two-site Ping Pong mechanism"). D'autres enzymes catalysent des mécanismes ping pong à n sites. Ces enzymes contiennent des coenzymes (exemples : biotine, acide lipoïque, 4-phosphopantétheine) liés par covalence. Ces coenzymes transfèrent d'un site à l'autre les groupements modifiés lors de la réaction enzymatique. Le transfert du groupe carboxyle est médié par la biotine qui peut passer d'un site à l'autre. En effet, la biotine est covalamment liée à la transcarboxylase via une liaison amide entre le groupe pentanoyl de la biotine et le groupe aminobutyl d'une lysine de l'enzyme.
Sur un site : le méthyl-malonyl-CoA transfère son groupe carboxyle à la biotine pour former la carboxy-biotine-transcarboxylase et le propionyl-CoA est dissocié de l'enzyme. Sur l'autre site : le pyruvate se fixe indépendamment. La carboxy-biotine y est transférée et le pyruvate est carboxylé en oxaloacétate qui se dissocie de l'enzyme. Les équations des vitesses sont identiques au mécanisme Ping Pong classique mais les profils d'inhibitions sont inversés. |
6. Mécanismes d'inhibition des systèmes à 2 substrats Voir un cours sur les inhibitions des réactions enzymatiques. |
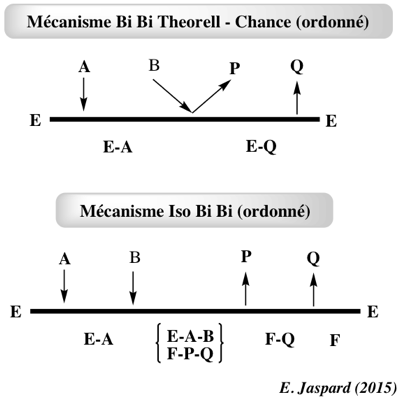
7. Mécanismes plus complexes a. Pas de complexe central ou isomèrisation de l'enzyme Mécanisme séquentiel Theorell - Chance (exemple : histone acétyltransférase p300) : le complexe central ne s'accumule pas car les substrats s'associent faiblement avec l'enzyme. En conséquence, la concentration à l'état stationnaire du complexe central est négligeable du point de vue cinétique. Exemple de mécanisme Iso Bi Bi Ordonné où l'enzyme libre s'isomèrise (forme E en forme F).
|
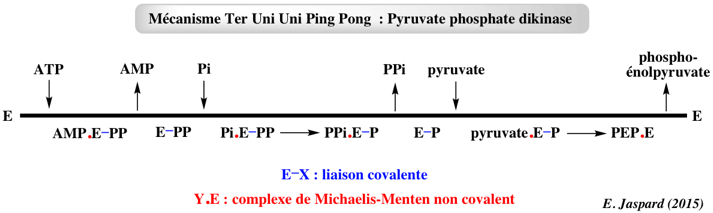
b. Enzymes avec plus de 2 substrats / plus de 2 produits Exemples d'enzymes qui catalysent des réactions avec 3 substrats et 2 ou 3 produits : glutamine synthétase adenylyltransferase (EC 2.7.7.42); les aminoacyl tRNA synthétases (EC 6.1.1.*); l'acétyl-CoA synthétase (EC 6.2.1.1); la succinyl-CoA synthétase (EC 6.2.1.5); les mono-oxygénases. Exemple de mécanisme Ter Uni Uni Ping Pong de la pyruvate phosphate dikinase (EC 2.7.9.1). Cette enzyme est utilisée par les plantes en C4.
|
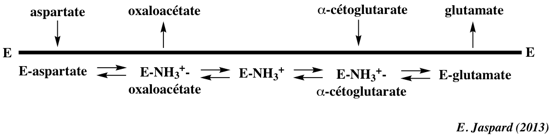
Autres exemples de mécanismes avec plus de 2 substrats / plus de 2 produits a. Le complexe multi-enzymatique mitochondrial α-cétoglutarate deshydrogénase (EC 1.2.4.2, EC 2.3.1.61 et EC 1.6.4.3) est un élément clé de la régulation du cycle de Krebs.
b. Figure ci-dessous :
|
Exemples d'enzymes avec 4 substrats et 3 ou 4 produits
|
| Nombre de mécanismes théoriques vs. nombre de mécanismes réels | |||
| Type de mécanismes | Nombre théorique de mécanismes | Nombre réel de mécanismes différents1 | Exemples |
| Ter Ter | 6 mécanismes : Ter Ter Ordonné / Bi Uni Uni Bi Ping Pong / Bi Bi Uni Uni Ping Pong / Uni Bi Bi Uni Ping Pong / Uni Uni Bi Bi Ping Pong / Hexa Uni Ping Pong |
synthèse de la S-adénosyl methionine |
|
| Quad2 Bi | 4 | 3 | mevalonic pyrophosphate décaxboxylase |
| Quad Ter | 10 | 5 | citrate-splitting enzyme |
| Quad Quad | 20 | 9 | carbamyl phosphate synthétase |
|
1. Différents modèles peuvent aboutir à des équations de vitesse formellement identiques. Exemples : le système Bi Bi au hasard analysé avec l'hypothèse du quasi - équilibre et le système Bi Bi ordonné analysé avec l'hypothèse de l'état stationnaire conduisent à la même expression de la vitesse de la réaction. 2. Pour une description détaillée des mécanismes et des équations d'un système Quad (4 substrats), voir : Yago et al. (2013) "Initial Rate Equations in Four-Substrate Enzyme Reactions. Application to Discriminate between Some Mechanisms and Evaluate Global Kinetic Parameters" MATCH Commun. Math. Comput. Chem. 69, 215-248 |
|||
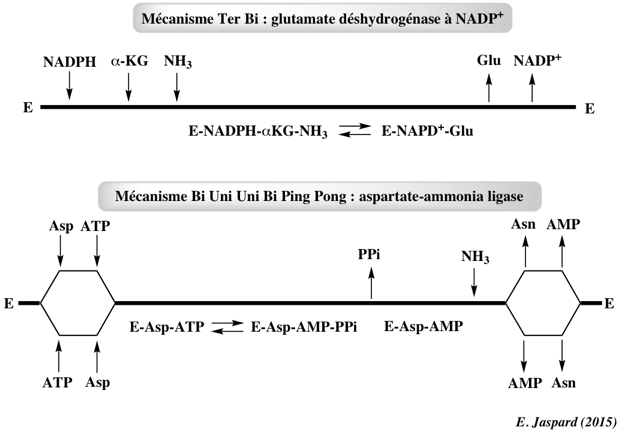
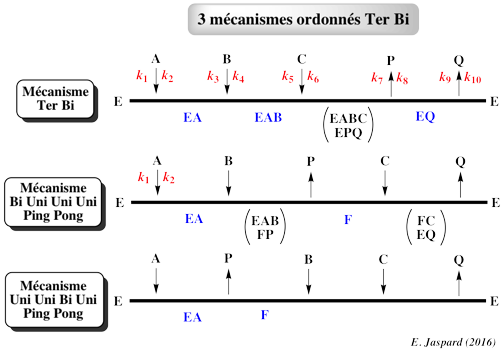
Ci-dessous : les 3 mécanismes ordonnés Ter Bi.
|
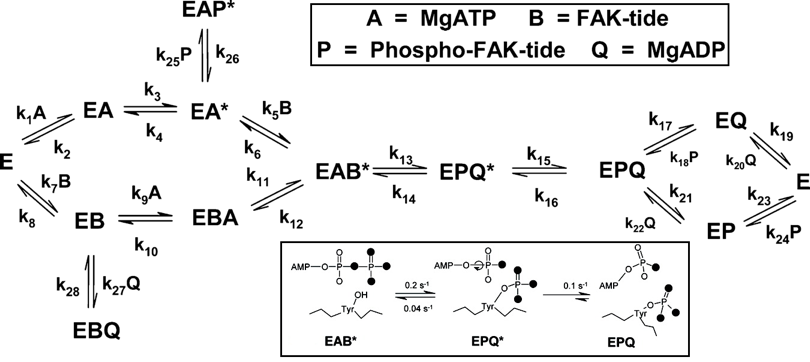
c. Etude de la protéine kinase FAK-1 - Schneck et al. (2010) FAK-1 : "Focal Adhesion Kinase-1" est une protéine kinase (tyrosine kinase non-récepteur) ubiquitaire dans toutes les cellules. Méthode de mesure des vitesses de catalyse : mesures de la radioactivité incorporée lors de la phosphorylation d'un substrat synthétique, le peptide Ac-RRRRRRSETDDYAEIID-NH2 (Y est le site de phosphorylation) appelé FAK-tide. FAK-1 est un homodimère (2 x 119 kDa). Son domaine catalytique contient un motif SH2, flanqué d'un domaine N-terminal FERM et d'un domaine C-terminal contenant la séquence de ciblage pour l'adhésion focale ("Focal Adhesion Targeting" - FAT). Sous sa forme inactive, le domaine FERM relie les extrémités des lobes N- et C-terminaux du domaine kinase et s'étend au travers du site actif, bloquant ainsi l'accès au substrat, donc la catalyse. La séquence du peptide FAK-tide correspond donc à la séquence du site d'autophosphorylation de FAK-1 (autophosphorylation de Tyr397) : Tyr397 n'est pas située dans la boucle d'activation mais dans une région qui relie domaine FERM et le domaine catalytique. |
| Hypothèses | Expressions des paramètres cinétiques |
Valeurs des paramètres cinétiques |
Tous les complexes binaires (enzyme-substrats, enzyme-produits et les deux complexes abortifs) sont en équilibre rapide. L'expression VMax / [E0] est décrite par trois étapes lentes (k13, k14 et k15) avec k3 et k11 >> k12, k13, k14, et k15 et k16 négligeables. |
kcat = 1 / [1/k3 + 1/k11 + (k14 + k15) / (k13 . k15 + 1/k15)] ≈ (k13 . k15) / (k13 + k14 + k15) |
kcat = 0.052 s-1 |
| KMMgATP = [k13 . k15 / (k13 + k14 + k15)] [(k10 + k11) / k9 . k11] | KMMgATP = 1.2 μM KMFAK-tide = 5.6 μM |
|
| KMFAK-tide = [k6 . (k14 + k15) . (1 + k3 / k4)] / [(k13 + k14 + k15) . (k3 . k5 / k4)] | KIMgATP = 1.3 μM KIFAK-tide = 6.1 μM |
| Profils d'inhibition par les produits et par les complexes abortifs | ||
| Mg-ATP | FAK-tide | |
| Mg-ADP | compétitif - KI = 1.4 μM | non compétitif |
| Phospho-FAK-tide | non compétitif | compétitif - KI = 220 μM |
| AMP-PNP [adénosine 5′-(β,γ-imido)triphosphate] |
compétitif - KI = 4.5 μM | non compétitif |
| Phe/Tyr-FAK-tide | non compétitif | compétitif - KI = 700 μM |
Deux complexes [substrat - produit] abortifs ("dead-end") sont possibles : E-MgADP-FAK-tide (EBQ) et E-MgATP-phospho-FAK-tide (EAP*). Le complexe E-MgADP-peptide (EBQ) a été observé pour d'autre kinases : exemple, le complexe E-MgADP-Ser-peptide de la protéine kinase AMPc-dépendante. |
||
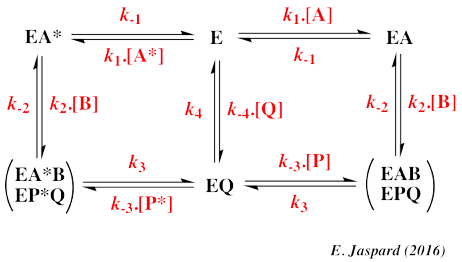
L'ensemble des résultats sont en faveur d'un mécanisme Bi-Bi au hasard (figure ci-dessous).
Source : Schneck et al. (2010) EAB* et EPQ* : E-MgATP-FAK-tide et E-MgADP-phospho-FAK-tide, respectivement, avec la boucle d'activation fermée EPQ : E-MgADP-phospho-FAK-tide avec la boucle d'activation ouverte L'étape de catalyse (kcat) est limitante à cause de deux évènements lents (encadré du bas de la figure ci-dessus) :
|
8. Utilisation d'isotopes pour distinguer les mécanismes : échange isotopique et effets des isotopes Voir un cours sur la radioactivité. Les hypothèses sur lesquelles reposent l'échange isotopique et les effets des isotopes ne peuvent pas être vraies simultanément d'où la nécessité d'ajuster les conditions expérimentales pour étudier l'un ou l'autre de ces deux processus. |
| Utilisations des isotopes pour les études cinétiques | ||
| Echange isotopique | Effets des isotopes | |
| localisation de la substitution des isotopes | éloigné du site actif | - située au niveau de la liaison impliquée dans la réaction : effet isotopique primaire |
| étendue de la substitution | à l'état de trace | - totale (100%) |
| propriété utilisée des isotopes | leur radioactivité | leur différence de masse |
| isotopes employés | 3H, 14C, 32P, 35S | 1H (protium), 2H (deutérium), 3H (tritium) |
| Voir : chapitre 7 - "Fundamentals of enzyme kinetics" (2001) A. Cornish-Bowden, Portland Press Ltd. | ||
a. Etude de l'échange isotopique L'hypothèse élémentaire est que la substitution d'un isotope par un autre n'a pas d'effet sur les propriétés chimiques et que les effets sur les propriétés cinétiques sont négligeables. Le schéma ci-dessous représente le transfert d'un atome radioactif (représenté par une astérisque) de A* à P* dans un mécanisme ordonné.
Cet échange d'atomes requière que A* se fixe à E : il ne peut donc avoir lieu que s'il y a une concentration suffisante de E. La réaction d'échange doit donc être inhibée par de fortes concentrations de A ou de Q puisqu'ils sont en compétition avec A* pour E. Les effets de B et P sont plus subtils :
Deux équations en fonction des concentrations [EA*] et [EA*B] avec l'hypothèse de l'état stationnaire :
Pour [P*] => 0 (condition de vitesse initiale) : [EA*B] = k1.k2[A*][B][E] / k-1.(k-2 + k3) + (k2.k3[B]) La vitesse initiale d'échange isotopique s'écrit : vi* = k3.[EA*B] = k1.k2.k3[A*][B][E] / k-1.(k-2 + k3) + (k2.k3[B]) Expression de [E] Le traitement de l'équation ci-dessus est simplifié si on fait l'hypothèse que les concentrations des substrats non marqués ne sont pas modifiées par la présence de traces de substrats radioactifs. De la même manière la concentration [E] est la même que s'il n'y avait pas de substrats radioactifs. Il y a deux manières de s'affranchir d'une expression complexe de [E] : Soit la vitesse d'échange isotopique est étudiée avec les réactions impliquant le substrat non marqués à l'équilibre : Soit on ne prend pas en compte les vitesses d'échange isotopique mais le rapport de ces vitesses d'échange. |
b. Etude des effets des isotopes A l'inverse de l'échange isotopique, l'hypothèse est qu'il y a une différence de propriétés à l'équilibre et de cinétiques entre les isotopes. L'utilisation d'isotopes permet, entre autre, de distinguer divers mécanismes enzymatiques. L'énergie de la liaison C-H en fonction de la distance séparant les deux atomes ne dépend que du nuage électronique qui les entoure et cette énergie est la même pour tous les isotopes de C et de H. Cependant, quand une liaison vibre, les niveaux d'énergies de cette liaison dépendent des masses des atomes en vibration : en conséquence, ces niveaux d'énergies sont différents pour des isotopes différents.
Il existe plusieurs méthodes pour étudier les effets des isotopes : a. La comparaison des représentations réciproques avec des substrats non marqués et marqués :
b. La "compétition interne" où les substrats non marqués et marqués sont présents en même temps. Le changement dans leur proportion reflète l'effet isotopique sur [VMax / KM] du substrat marqué. Cette méthode est utilisée avec 3H ou 14C ou avec l'abondance naturelle de 13C, 15N et 18O. c. L'étude des perturbations de l'équilibre quand un substrat marqué et le produit correspondant non marqué sont présents à l'équilibre. |
| 10. Liens Internet et références bibliographiques |
|
King E.L. & Altman C. (1956) "A Schematic Method of Deriving the Rate Laws for Enzyme-Catalyzed Reactions" J. Phys. Chem. 60, 1375 - 1378 Cleland W.W. (1963) "The kinetics of enzyme-catalyzed reactions with two or more substrates or products. I. Nomenclature and rate equations" Biochim. Biophys. Acta. 67, 104-137 Elliott & Tipton (1974) "A kinetic analysis of enzyme systems involving four substrates" Biochem. J. 141, 789 - 805 Segel I.H. (1975) "Enzyme Kinetics", John Wiley, New York Cornish-Bowden A. (1977) "An automatic method for deriving steady-state rate equations" Biochem. J. 165, 55 - 59 Viratelle O. (1993) "Protéines et enzymes - TD" - Collection Méthodes, Hermann - ISBN : 2-7056-6185-9 "Fundamentals of enzyme kinetics" (2001) A. Cornish-Bowden, Portland Press Ltd. - ISBN : 1 85578 070 0 |
|
|
Cleland W.W. (2003) "The use of isotope effects to determine enzyme mechanisms" J. Biol. Chem. 278, 51975 - 51984 Borra et al. (2004) "Substrate specificity and kinetic mechanism of the Sir2 family of NAD+-dependent histone/protein deacetylases" Biochemistry 43, 9877 - 9887 Bartling & Raetz (2008) "Steady-State Kinetics and Mechanism of LpxD, the N-Acyltransferase of Lipid A Biosynthesis" Biochemistry 47, 5290 - 5302 |
|
|
Qi et al. (2009) "Generating rate equations for complex enzyme systems by a computer-assisted systematic method" BMC Bioinformatics 10, 238 Schneck et al. (2010) "Kinetic Mechanism and Rate-Limiting Steps of Focal Adhesion Kinase-1" Biochemistry 49, 7151 - 7163 Qi et al. (2011) "Detailed kinetics and regulation of mammalian 2-oxoglutarate dehydrogenase" BMC Biochem. 12, 53 |
|
|
Al-Haque et al. (2012) "A robust methodology for kinetic model parameter estimation for biocatalytic reactions" Biotechnol Prog. 28, 1186 - 1196 Masoudi et al. (2013) "Chasing acyl carrier protein through a catalytic cycle of lipid A production" Nature 505, 422 - 426 Beard & Wilson (2014) "Structure and Mechanism of DNA Polymerase β" Biochemistry 53, 2768 - 2780 |
|
![]()